Toxin from brain cells triggers neuron loss in human ALS model
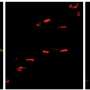
 release a toxin that kills motor neurons via a recently discovered process described as a \"controlled cellular explosion.\" Credit: Diane Re, Columbia University Medical Center")
In most cases of amyotrophic lateral sclerosis (ALS), or Lou Gehrig's disease, a toxin released by cells that normally nurture neurons in the brain and spinal cord can trigger loss of the nerve cells affected in the disease, Columbia researchers reported today in the online edition of the journal Neuron.
The toxin is produced by star-shaped cells called astrocytes and kills nearby motor neurons. In ALS, the death of motor neurons causes a loss of control over muscles required for movement, breathing, and swallowing. Paralysis and death usually occur within 3 years of the appearance of first symptoms.
The report follows the researchers' previous study, which found similar results in mice with a rare, genetic form of the disease, as well as in a separate study from another group that used astrocytes derived from patient neural progenitor cells. The current study shows that the toxins are also present in astrocytes taken directly from ALS patients.
"I think this is probably the best evidence we can get that what we see in mouse models of the disease is also happening in human patients," said the study's senior author, Serge Przedborski, MD, PhD, the Page and William Black Professor of Neurology (in Pathology and Cell Biology), Vice Chair for Research in the Department of Neurology, and co-director of Columbia's Motor Neuron Center.
The findings also are significant because they apply to the most common form of ALS, which affects about 90 percent of patients. Scientists do not know why ALS develops in these patients; the other 10 percent of patients carry one of 27 genes known to cause the disease.
"Now that we know that the toxin is common to most patients, it gives us an impetus to track down this factor and learn how it kills the motor neurons," Dr. Przedborski said. "Its identification has the potential to reveal new ways to slow down or stop the destruction of the motor neurons."
. The right side of the image shows a healthy motor neuron on top of astrocytes from people unaffected by ALS. Credit: Diane Re, Columbia University Medical Center")
In the study, Dr. Przedborski and study co-authors Diane Re, PhD, and Virginia Le Verche, PhD, associate research scientists, removed astrocytes from the brain and spinal cords of six ALS patients shortly after death and placed the cells in petri dishes next to healthy motor neurons. Because motor neurons cannot be removed from human subjects, they had been generated from human embryonic stem cells in the Project A.L.S./Jenifer Estess Laboratory for Stem Cell Research, also at CUMC.
Within two weeks, many of the motor neurons had shrunk and their cell membranes had disintegrated; about half of the motor neurons in the dish had died. Astrocytes removed from people who died from causes other than ALS had no effect on the motor neurons. Nor did other types of cells taken from ALS patients.
The researchers confirmed that the cause of the motor neurons' death was a toxin released into the environment by immersing healthy motor neurons in the astrocytes' culture media. The presence of the media, even without astrocytes, killed the motor neurons.
How the Toxin Triggers Motor Neuron Death
The researchers have not yet identified the toxin released by the astrocytes. But they did discover the nature of the neuronal death process triggered by the toxin.The toxin triggers a biochemical cascade in the motor neurons that essentially causes them to undergo a controlled cellular explosion.
Drs. Przedborski, Re, and Le Verche found that they could prevent astrocyte-triggeredmotor neuron death by inhibiting one of the key components of this molecular cascade.
These findings may lead to a way to prevent motor neuron death in patients and potentially prolong life. But the therapeutic potential of such inhibition is far from clear. "For example, we don't know if this would leave patients with living but dysfunctional neurons," Dr. Przedborski said. The researchers are now testing the idea of inhibition in animal models of ALS.
New Human Cell Model of ALS Will Speed Identification of Potential Therapies
The development of new therapies for ALS has been disappointing, with more than 30 clinical trials ending with no new treatments since the 1995 FDA approval of riluzole.
The lack of progress may be partly because animal models used to study ALS do not completely recreate the human disease. The new all-human cell model of ALS created for the current study may improve scientists' ability to identify useful drug targets, particularly for the most common form of the disease.
"Although there are many neurodegenerative disorders, only for a handful do we have access to a simplified model that is relevant to the disease and can therefore potentially be used for high-throughput drug screening. So this model is quite special," Dr. Przedborski said. "Here we have a spontaneous disease phenotype triggered by the relevant tissue that causes human illness. That's one important thing. The other important thing is that this model is derived entirely from human elements. This is probably the closest, most natural model of human ALS that we can get in a dish."
More information: The paper is titled "Necroptosis drives motor neuron death in models of both sporadic and familial ALS."