Throwing a loop to silence gene expression
All human cells contain essentially the same DNA sequence – their genetic information. How is it possible that shapes and functions of cells in the different parts of the body are so different? While every cell's DNA contains the same construction master plan, an additional regulatory layer exists that determines which of the many possible DNA programs are active. This mechanism involves modifications of genome-bound histone proteins or the DNA itself with small chemical groups (e.g. methylation). It acts on top of the genetic information and is thus called 'epi'-genetic from the corresponding Greek word that means 'above' or 'attached to'.
"Epigenetics has fundamentally changed our view on how the genetic information is used", says Dr. Karsten Rippe from the German Cancer Research Center, who is studying this process with his team. "Epigenetic modifications can be rapidly set or removed to reversibly change cell function. At the same time, epigenetic patterns can be stably inherited through cell division and possibly also to the next generation."
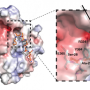
It turns out that deciphering the cell's 'epigenetic code' is a challenging task: Hundreds of proteins in the cell are linked in large networks to 'write', 'erase' or 'read' about 140 different chemical modifications of histone proteins and DNA that have been identified so far. Understanding how epigenetic regulation operates for a specific part of the genome thus requires an integrative approach that considers the connections between different factors. Accordingly, the researchers, together with their colleagues from the DKFZ and the LMU Munich, conducted a comprehensive analysis of a prototypic epigenetic network. They studied how certain DNA sequences were silenced by histone and DNA methylation that would make the genome instable if active and would thus favor cancer development.
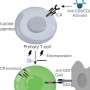
Based on maps of epigenetic signals and interactions of proteins with the genome, they developed a mathematical model for epigenetic silencing. "The silencing mechanism we found works much like throwing a loop with a lasso to catch something", says Katharina Müller-Ott, the first author of the study: "Several factors bind the silencing enzyme stably to certain sites in the genome. Because the DNA randomly moves around and forms transient loops, the enzyme hits other regions in the genome nearby, which then become modified and are switched off."
By virtue of their quantitative description of this process, the researchers were able to predict how the silencing network would react in response to perturbations like changes of the abundance of proteins or the activity of the enzymes involved. The scientists in the groups of Karsten Rippe and Thomas Höfer at the DKFZ are now continuing to further develop and apply their model to deregulated epigenetic signaling in leukemia. By evaluating genome-wide maps of epigenetic signals with mathematical models they are identifying tumor-specific changes in cell samples from patients with blood cancer. Furthermore, they are dissecting how epigenetic signals can be used to predict therapy response and how drugs affect the epigenetic program.
More information: Müller-Ott, K., Erdel, F., Matveeva, A., Hahn, M., Mallm, J.-P., Rademacher, A., Bauer, C., Zhang, Q., Kaltofen, S., Schotta, G., Höfer, T. & Rippe, K. (2014). Specificity, propagation and memory of pericentric heterochromatin. Mol. Syst. Biol. 2014, DOI: 10.15252/msb.20145377