Multiple models reveal new genetic links in autism

With the help of mouse models, induced pluripotent stem cells (iPSCs) and the "tooth fairy," researchers at the University of California, San Diego School of Medicine have implicated a new gene in idiopathic or non-syndromic autism. The gene is associated with Rett syndrome, a syndromic form of autism, suggesting that different types of autism spectrum disorder (ASD) may share similar molecular pathways.
The findings are published in the Nov. 11, 2014 online issue of Molecular Psychiatry.
"I see this research as an example of what can be done for cases of non-syndromic autism, which lack a definitive group of identifying symptoms or characteristics," said principal investigator Alysson Muotri, PhD, associate professor in the UC San Diego departments of Pediatrics and Cellular and Molecular Medicine. "One can take advantage of genomics to map all mutant genes in the patient and then use their own iPSCs to measure the impact of these mutations in relevant cell types. Moreover, the study of brain cells derived from these iPSCs can reveal potential therapeutic drugs tailored to the individual. It is the rise of personalized medicine for mental/neurological disorders."
But to effectively exploit iPSCs as a diagnostic tool, Muotri said researchers "need to compare neurons derived from hundreds or thousands of other autistic individuals." Enter the "Tooth Fairy Project," in which parents are encouraged TO register for a "Fairy Tooth Kit," which involves sending researchers like Muotri a discarded baby tooth from their autistic child. Scientists extract dental pulp cells from the tooth and differentiate them into iPSC-derived neurons for study.
"There is an interesting story behind every single tooth that arrives in the lab," said Muotri.
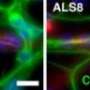
The latest findings, in fact, are the result of Muotri's first tooth fairy donor. He and colleagues identified a de novo or new disruption in one of the two copies of the TRPC6 gene in iPSC-derived neurons of a non-syndromic autistic child. They confirmed with mouse models that mutations in TRPC6 resulted in altered neuronal development, morphology and function. They also noted that the damaging effects of reduced TRPC6 could be rectified with a treatment of hyperforin, a TRPC6-specific agonist that acts by stimulating the functional TRPC6 in neurons, suggesting a potential drug therapy for some ASD patients.
The researchers also found that MeCP2 levels affect TRPC6 expression. Mutations in the gene MeCP2, which encodes for a protein vital to the normal function of nerve cells, cause Rett syndrome, revealing common pathways among ASD.
"Taken together, these findings suggest that TRPC6 is a novel predisposing gene for ASD that may act in a multiple-hit model," Muotri said. "This is the first study to use iPSC-derived human neurons to model non-syndromic ASD and illustrate the potential of modeling genetically complex sporadic diseases using such cells."