Rare mutation causes vitamin A deficiency and eye deformities

Researchers at the University of Michigan and UC Davis have solved a genetic mystery that has afflicted three unrelated families, and possibly others, for generations. These families have been plagued by a variety of congenital eye malformations, including small eyes with poor vision and the complete absence of eyes. But until now, no one could figure out the genetic basis for these conditions.
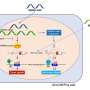
By mapping and sequencing family DNA, the research team found mutations in a protein (RBP4) that transports a form of vitamin A called retinol, an essential nutrient for eye development. The mutations create a functional "double-whammy."
First, the mutated proteins fail to transport retinol to the developing embryo. Then, to make matters worse, they also block the cell surface receptor for RBP4 (called STRA6), keeping healthy proteins from delivering their nutritional payload. The end result is a severe retinol deficiency and subsequent birth defects. The research appears online April 23 in the journal Cell.
"Instead of being inactive, the mutated proteins have altered function," said senior author Tom Glaser, a professor in the Department of Cell Biology and Human Anatomy at UC Davis who began this project at the University of Michigan. "They fail to bind to retinol, but they also plug up the receptor, binding 40 times more tightly than the unmutated protein. The mutants act like goalies, keeping retinol away from the receptors."
The paper also highlights a unique collaboration between families, clinicians and researchers, according to Christine Nelson, a senior clinical author of the study and professor in the Department of Ophthalmology and Visual Sciences at the University of Michigan's Kellogg Eye Center. The study arose from Nelson's discovery that two of her young patients with similar eye malformations were related. The project gained momentum when the team received an unexpected "gift" from the family patriarch—an invaluable family tree that, together with old photographs, revealed a new type of inheritance.
"Some family members only took pictures from one side, or tilted their heads a certain way so they could see better," Nelson said. "This visual evidence helped us identify eye defects in earlier generations."
RBP4 is secreted by the liver and plays a critical role in eye development. Because retinol is not water-soluble, it needs a vehicle to carry it in the bloodstream to the STRA6 receptor. The transaction becomes even more intricate in utero, as the nutrient must be passed through the placenta from mother to baby.
This complicated transfer process plays a crucial role in how the defect is inherited from one generation to the next. The largest family in the study had endured eye issues for five generations. However, though the mutated gene is dominant, it only causes problems when passed through the mother, often skipping generations. One child inherited the gene through the father and exhibited the trait, but experienced only mild symptoms. The research team had inadvertently discovered a unique form of maternal inheritance.
"When the trait is inherited from the mother, there's a problem transporting retinol from the mother's liver to the placenta and from the placenta to the fetus," said Glaser. "It's a two-part process and the retinol has to be handed off, like a relay race, to cross between maternal and fetal circulation. When inherited from the father, only one leg of the journey is defective, allowing enough vitamin A to get through to prevent the birth defect."
This unusual maternal inheritance pattern may have implications for other families experiencing congenital diseases. However, the research is already having profound implications for these particular families. In some cases, mothers and babies have both carried the mutation but experienced no ocular birth defects, showing the defective pathway can be circumvented.
"While further clinical research is needed, it appears that we might be able to save a child from blindness with a simple and inexpensive treatment - an extra vitamin A pill," Nelson said. "This supplementation relies on an alternate pathway, independent of RBP, that delivers another form of vitamin A, called retinyl ester, bypassing the mutations altogether.
Nelson suggests that women with this family history of eye malformations or those who learn that they carry the mutation consult with their obstetricians about taking vitamin A supplements during pregnancy. The discussion should begin before pregnancy since major steps in eye development take place in the first two months of gestation. Thus, a woman might not know she is pregnant when treatment is optimal.
"We're lucky that nature has created this parallel pathway because vitamin A is so important to a developing fetus," said Christopher Chou, the study's first author and a resident physician in emergency medicine at the University of Michigan. "It's not often you have a genetic defect on which you can directly intervene. This study illustrates how understanding one's genetic make-up can add a personal touch to medical care."
The researchers still have much work to do. Glaser notes the team would like to map the entire genetic pathway associated with eye malformations. In addition, the study could help researchers investigate other genetic conditions.
"We need to look at genes governing vitamin A transport, as well as transport genes that affect the development of other organs," Nelson said. "Also, there may be additional diseases that result from defective placental transport."