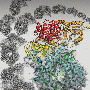Possible path toward first anti-MERS drugs
If you haven't heard of Middle East Respiratory Syndrome, thank geography, NGOs, and government agencies like the Centers for Disease Control and Prevention and the World Health Organization (WHO) for keeping the new disease in check. In camels, which act as a reservoir for the causative coronavirus, the illness causes a runny nose; in humans, it causes a cough, fever, and, in 36 percent of cases, death, according to the WHO.
The virus has recently spread to South Korea, but the first cases were reported in Saudi Arabia in 2012. Vaccinating camels has been proposed as a solution, but a suitable animal vaccine has yet to be developed, and there are currently no drug treatments available for the virus.
This week at the American Crystallographic Association (ACA) 2015 Meeting in Philadelphia, researchers from the National Cancer Institute will present three newly determined crystal structures, which collectively provide a starting point for structure-based drug design efforts to combat MERS. The work will be presented by George Lountos, a scientist in the lab of primary investigator David Waugh.
MERS-CoV is part of the betacoronavirus genus, whose infamous cousins include the viruses responsible for SARS and the common cold. As a positive-sense species, its viral RNA is laid out in the 5' to 3' direction of RNA translation, allowing it to skip transcription to messenger RNA and be directly translated into proteins.
When the virus initiates replication, its genome gets translated into two large polyprotein chains, which are then cleaved into smaller functional proteins by enzymes called proteases. At the 5' end, a long open reading frame encodes for 16 non-structural proteins, one of which is the 3C-like protease, or 3CLpro. Once active, 3CLpro cleaves the other non-structural proteins to form mature proteins that are involved in viral replication. By blocking its activity, you can completely shut the process down.
Inhibiting viral replication by blocking proteases is a tried and true method of treating infections and has been proven to save lives of people with other types of viral infections, like HIV.
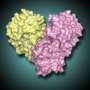
Using X-ray crystallography, the researchers determined three crystal structures of a catalytically inactive variant, C148A, of 3CLpro - one of the free enzyme, and two representing enzyme-product complexes.
To find suitable drug development candidates, the researchers intend to use fragment-based screening, in which low weight molecular compounds with weak affinities for the protease are soaked into its crystallized protein form. The fragments are typically lighter than 250 Daltons—for comparison, a caffeine molecule weighs 194 Daltons—and are selected in advance for favorable drug-like properties and amenability to further chemical modifications. Structural information gleaned from the fragments bound to the 3CLpro can then be used to optimize the fragments into higher affinity inhibitors.
Drug design efforts from other laboratories have focused on peptidomimetics, small protein-like chains that mimic peptides. While these are easily synthesized, they are not easily administered orally, which limits their appeal as antivirals.
Additionally, the researchers have identified an enzyme-product complex within their current crystal structures. When crystallized, the enzyme forms homodimers, or molecules composed of paired identical proteins. The 3CLpro can cleave itself from the long polyprotein chain by recognizing cleavage site sequences that flank it, which resulted in the discovery of a 3CLpro molecule bound to the 3CLpro homodimer active site via its autoprocessed C-terminal tail in the crystal lattice. This gives the researchers a snapshot of how the 3CL-protease can recognize the cleavage site of an unprocessed protein chain, and thus crucial information about which areas of the enzyme's active site they need to target to block the interaction.
Lountos and his colleagues have recently begun working through their library of compounds, and they estimate it will take at least a year to sort through the data from the bound fragments and find a lead compound, upon which further biochemistry studies will then need to be done before the longer process of drug design can begin.