Targeting glucose production in liver may lead to new diabetes therapies
High blood sugar is a defining characteristic of Type 2 diabetes and the cause of many of the condition's complications, including kidney failure, heart disease, and blindness. Most diabetes medications aim to maintain normal blood sugar (glucose) levels and prevent high blood sugar by controlling insulin.
A new University of Iowa study shows that another biological checkpoint, known as the Mitochondrial Pyruvate Carrier (MPC), is critical for controlling glucose production in the liver and could potentially be a new target for drugs to treat diabetes.
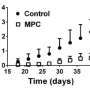
The study, led by Eric Taylor, PhD, UI assistant professor of biochemistry, and published Sept. 3 in the journal Cell Metabolism, shows that disabling MPC reduces blood sugar levels in mouse models of Type 2 diabetes.
Glucose is primarily made in the liver and requires the molecular building blocks to pass through specialized cellular compartments called mitochondria. Mitochondria use a small molecule called pyruvate as the starting point for synthesizing glucose, and the pyruvate is imported into the mitochondria through the MPC portal.
Taylor and his colleagues showed that disabling the MPC in mouse livers shuts down this major route of glucose production. However, because glucose is a critical cellular fuel, there are "back-up" mechanisms. When the MPC was disabled in mouse livers, another glucose-producing mechanism was activated to compensate. This alternative mechanism uses molecules from protein as the building blocks for glucose.
"Essentially, what we found is that disruption of the MPC makes the liver less efficient at making glucose and, as a result, the liver burns more fat for energy, makes less cholesterol, and makes less glucose in models of diabetes," explains Taylor, who also is a member of the Fraternal Order of Eagles Diabetes Research Center at the UI. "This overall change in metabolism matches outcomes that would be therapeutically desirable for people with diabetes."
The new research is based on earlier work by Taylor's group and others, which identified the genes for MPC. In the new study, the UI researchers use this genetic information to specifically disrupt MPC activity in animal models. They found that disrupting MPC in normal mice doesn't cause low blood sugar, or hypoglycemia, which would be important for the safety of any new treatment targeting MPC. In mouse models of Type 2 diabetes, however, loss of the MPC activity in the liver decreases high blood sugar and improves glucose tolerance. The study also suggests that MPC activity contributes to excess glucose production and high blood sugar levels in Type 2 diabetes.
The therapeutic potential of targeting glucose production in the liver is supported by the fact that metformin, the most widely used and staple treatment for Type 2 diabetes, also decreases glucose synthesis in the liver by disrupting mitochondrial metabolism, although the exact mechanisms underlying this drug's action on mitochondria are controversial.
However, Taylor cautions that additional research will be required to determine if inhibiting MPC activity might be a safe approach for human therapies, especially in people under high levels of physical stress or with other medical complications.
The team plans to extend their studies to cultured human liver cells to determine if disabling the MPC produces the same metabolic effects as seen in the mouse studies, and to make sure that inhibiting this checkpoint does not produce dangerous side effects.
More information: Hepatic Mitochondrial Pyruvate Carrier 1 Is Required for Efficient Regulation of Gluconeogenesis and Whole-Body Glucose Homeostasis, DOI: dx.doi.org/10.1016/j.cmet.2015.07.027 . www.cell.com/cell-metabolism/a … 1550-4131(15)00390-3