Genetic study provides first-ever insight into biological origin of schizophrenia
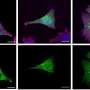
 located at the synapses of primary human neurons. Credit: Heather de Rivera (McCarroll lab).")
A landmark study, based on genetic analysis of nearly 65,000 people, has revealed that a person's risk of schizophrenia is increased if they inherit specific variants in a gene related to "synaptic pruning"—the elimination of connections between neurons. The findings represent the first time that the origin of this devastating psychiatric disease has been causally linked to specific gene variants and a biological process. They also help explain decades-old observations: synaptic pruning is particularly active during adolescence, which is the typical period of onset for schizophrenia symptoms, and brains of schizophrenic patients tend to show fewer connections between neurons. The gene, called complement component 4 (C4), plays a well-known role in the immune system but has now been shown to also play a key role in brain development and schizophrenia risk. The insight may allow future therapeutic strategies to be directed at the disorder's roots, rather than just its symptoms.
The study, which appears online in the Jan. 27, 2016 issue of Nature, was led by researchers from the Broad Institute's Stanley Center for Psychiatric Research, Harvard Medical School, and Boston Children's Hospital. They include senior author Steven McCarroll, director of genetics for the Stanley Center and an associate professor of genetics at Harvard Medical School; Beth Stevens, a neuroscientist and assistant professor of neurology at Boston Children's Hospital and institute member at the Broad; Michael Carroll, a professor at Harvard Medical School and researcher at Children's Hospital; and first author Aswin Sekar, an M.D./Ph.D. student at Harvard Medical School.
The study has the potential to reinvigorate translational research on a debilitating disease. Schizophrenia is a devastating psychiatric disorder that afflicts approximately one percent of the population and is characterized by hallucinations, emotional withdrawal, and a decline in cognitive function. These symptoms most frequently begin in patients when they are teenagers or young adults. First described more than 130 years ago, schizophrenia lacks highly effective treatments and has seen few biological or medical breakthroughs over the past half-century. In summer 2014, an international consortium, led by researchers at the Broad Institute's Stanley Center, identified more than 100 regions in the human genome that carry risk factors for schizophrenia. The newly published study now reports the discovery of the specific gene underlying the strongest of these risk factors and links it to a specific biological process in the brain.
"Since schizophrenia was first described over a century ago, its underlying biology has been a black box, in part because it has been virtually impossible to model the disorder in cells or animals," said McCarroll. "The human genome is providing a powerful new way in to this disease. Understanding these genetic effects on risk is a way of prying open that black box, peering inside, and starting to see actual biological mechanisms."
"This study marks a crucial turning point in the fight against mental illness," said Bruce Cuthbert, acting director of the National Institute of Mental Health. "Because the molecular origins of psychiatric diseases are little-understood, efforts by pharmaceutical companies to pursue new therapeutics are few and far between. This study changes the game. Thanks to this genetic breakthrough we can finally see the potential for clinical tests, early detection, new treatments, and even prevention."
The path to discovery
The remarkable story of discovery involved the collection of DNA from more than 100,000 people, detailed analysis of complex genetic variation in more than 65,000 human genomes, development of an innovative analytical strategy, examination of postmortem brain samples from hundreds of people, and the use of animal models to show that a protein from the immune system also plays a previously unsuspected role in the brain.

Worldwide search for data finds a clue; new research solves the mystery
Over the past five years, geneticists led by the Broad Institute's Stanley Center for Psychiatric Research and its collaborators around the world collected more than 100,000 human DNA samples from 30 different countries to locate regions of the human genome harboring genetic variants that increase the risk of schizophrenia. The strongest signal by far was on chromosome 6, in a region of DNA long associated with infectious disease, causing some observers to suggest that schizophrenia might be triggered by an infectious agent. But researchers had no idea which of the hundreds of genes in the region was actually responsible or how it acted.
Based on analyses of the genetic data, McCarroll and Sekar focused on a region containing an unusual gene called complement component 4 (C4). Unlike most genes, C4 has a high degree of structural variability: different people have different numbers of copies and different types of the gene. McCarroll and Sekar developed a new molecular technique to characterize the C4 gene structure in human DNA samples. They also measured C4 gene activity in nearly 700 post-mortem brain samples. They found that the C4 gene structure (DNA) could predict the C4 gene activity (RNA) in each person's brain - and used this information to infer C4 gene activity from genome data for 65,000 people with and without schizophrenia. These data revealed a striking correlation: patients who had particular structural forms of the C4 gene showed higher expression of that gene and, in turn, had a higher risk of developing schizophrenia.
Connecting cause and effect through neuroscience
But how exactly does C4—a protein known to mark infectious microbes for destruction by immune cells—affect the risk of schizophrenia?
Answering this question required synthesizing genetics and neurobiology. Beth Stevens, recent recipient of the MacArthur "Genius Grant," had found that other complement proteins in the immune system also played a role in brain development by studying an experimental model of synaptic pruning in the mouse visual system. Michael Carroll had long studied C4 for its role in immune disease, and developed mice with different numbers of copies of C4. The three labs set out to study the role of C4 in the brain.
They found that C4 played a key role in pruning synapses during maturation of the brain. In particular, they found that C4 was necessary for another protein (a complement component called C3) to be deposited onto synapses, as a signal that the synapses should be pruned. The data also suggested that the more C4 activity an animal had, the more synapses were eliminated in its brain at a key time in development.
The findings may help explain the longstanding mystery of why brains from people with schizophrenia tend to have a thinner cerebral cortex with fewer synapses than unaffected individuals do. The work may also help to explain why the onset of schizophrenia symptoms tends to occur in late adolescence: the human brain normally undergoes widespread synapse pruning during adolescence, especially in the cerebral cortex (the brain's outer layer, responsible for many aspects of cognition). Excessive synaptic pruning during adolescence and early adulthood, due to increased complement (C4) activity, could lead to the cognitive symptoms seen in schizophrenia.
"Once we had the genetic findings in front of us we started thinking about the possibility that complement molecules are excessively tagging synapses in the developing brain," Stevens said. "This discovery enriches our understanding of the complement system in brain development and in disease, and we could not have made that leap without the genetics. We're far from having a treatment based on this, but it's exciting to think that one day, we might be able to turn down the pruning process in some individuals and decrease their risk."
Opening a path toward early detection and potential therapies
Beyond providing the first insights into the biological origins of schizophrenia, the work raises the possibility that therapies might someday be developed that could "turn down" the level of synaptic pruning in individuals who show early symptoms of schizophrenia. This would be a dramatically different approach from current medical therapies, which address only a specific symptom of schizophrenia (psychosis) rather than the disorder's root causes, and which do not stop cognitive decline or other symptoms of the illness. The researchers emphasize that therapies based on these findings are still years down the road. Still, the fact that much is already known about the role of complement proteins in the immune system means that researchers can tap into a wealth of existing knowledge to identify possible therapeutic approaches. For example, anti-complement drugs are already under development for treating other diseases.
"For the first time, the origin of schizophrenia is no longer a complete black box," said Eric Lander, director of the Broad Institute. "While it's still early days, we've seen the power of understanding the biological mechanism of disease in other settings. Early discoveries about the biological mechanisms of cancer have led to many new treatments and hundreds of additional drug candidates in development. Understanding schizophrenia will similarly accelerate progress against this devastating disease that strikes young people."
The success of this effort was enabled by catalytic funding from the Stanley Center for Psychiatric Research at the Broad Institute and this paper has been dedicated to the late Ted Stanley. "Through philanthropy, we have been able to take bets on risky science with potentially transformative results," said Stanley Center Director Steven Hyman. "With support from the late Ted and Vada Stanley, Broad scientists have the freedom to bring together people, capabilities, and resources in innovative ways, at an unprecedented pace."
"In this area of science, our dream has been to find disease mechanisms that lead to new kinds of treatments," said McCarroll. "These results show that it is possible to go from genetic data to a new way of thinking about how a disease develops—something that has been greatly needed."
More information: Nature, nature.com/articles/doi:10.1038/nature16549