Putting the brakes on accelerated aging

Telomeres are to chromosomes what aglets are to shoelaces—protective caps that stop the tightly wound strings from unraveling. Every time a cell divides, its telomeres are trimmed, eventually becoming so short that the cell goes into an unproductive state of senescence. Lifestyle changes can slow or hasten this biological clock, which has become a popular area of research in the last few years. Scientists are looking at the link between aging and telomeres from all angles, from genetics to diet and lifestyle choices.
Researchers at the A*STAR Institute of Medical Biology (IMB) are examining another factor—the presence of a mutant protein known as progerin. The protein is produced by children with a rare genetic disease that causes premature aging, called Hutchinson-Gilford progeria syndrome. "The length of the telomeres in these children is about the same as that of an 80 or 90 year old," says Colin Stewart, a lead researcher in the study. "Somehow, progerin is accelerating this aspect of the aging process."
"This study offers one of the best pieces of evidence that damaged telomeres and premature senescence is detrimental to the body," says Oliver Dreesen, another lead researcher, discussing the work's implications for aging in general. "Maintaining the integrity of your telomeres is extremely important."
But probing further, the A*STAR team discovered something even more significant. By introducing to the mutant cells a specific protein found in the cell nucleus, telomere damage was prevented and the cells saved from early aging. "That really blew me away," says Dreesen.
Telomere link
Hutchinson-Gilford progeria affects one in every four million babies. Children with the disease typically show signs of aging from their first birthday, are bald by their second or third year, and die in their teens of heart failure or stroke. They are thinner and more fragile than their contemporaries, but remain mentally astute.
Early in his career, Stewart developed a keen interest in the new genetic manipulation techniques being used to create mouse models of various diseases. He focused on a fibrous protein found inside the nuclear lamina called lamin-A. "I became very interested in why this one rather boring protein in the nucleus, when mutated in different ways, leads to so many different types of disease." One of those diseases happened to be progeria, in which a single mutation in the gene that codes for lamin-A produces the mutant progerin. In 2003 Stewart produced the first mouse model of the premature aging syndrome. A few years later he moved to Singapore to develop new 'disease-in-a-dish' models of progeria using human embryonic stem cell technologies, which were advancing rapidly in the country.
Oliver Dreesen moved to A*STAR in 2009, having spent the last few years studying telomeres in a parasite that causes sleeping sickness. "One of the first discussions I had when I came to Singapore was with Colin, who told me about progeria," says Dreesen. "And I said to him: we have to look at telomere length." By that time, researchers had already established that telomeres played a role in the normal aging process. But those working on progeria were more preoccupied with the morphed structure of the cell nucleus, which projects like tiny bubbles from the original oval.
Together, Dreesen and Stewart developed a model of progeria using connective tissue cells known as fibroblasts in which they could gradually increase the dose of progerin. When progerin levels reached about 30 to 40 per cent of normal lamin-A levels, the cells began to exhibit some strange and pathological behavior: the nuclei lost its original shape, the telomeres showed signs of damage and the cell went into a state of senescence.
"They just sit there," says Dreesen to describe the irreversible growth arrest. "They are not dividing but are metabolically active and start to secrete all sorts of junk that breaks down the extracellular matrix." Dreesen and Stewart had published a study in 2013 in which they identified a specific biomarker for senescence that made these cells easy to detect.
Different types of cells accumulate progerin at different rates, which means that they arrive at this dangerous threshold sooner (as with blood vessel cells) or later (as with neural cells). The findings explained why children with progeria were more likely to die of cardiovascular diseases, while their mental capacity appeared unaffected. "Children who have progeria typically die from a heart attack or a stroke, which is associated with accelerated calcification of the blood vessels and also some types of atherosclerosis," says Stewart, describing, at the same time, a common cause of death worldwide. "Progeria may give us insights into how the normal vascular system ages."
To the rescue
Dreesen and Stewart then tried to see if they could prevent these progerin-induced cellular deteriorations. Their first candidate was an enzyme known to lengthen telomeres, called telomerase, which they introduced to the cells. To their surprise, telomerase prevented most of the deviant behavior of progerin-expressing cells, but it did not explain how it led to such detrimental outcomes.
To address this question they sought the aid of a colleague, Brian Burke, also at the IMB, who had developed a 'BioID' technique for studying how proteins interact. The technique involves tagging a protein of interest—in this case lamin-A or the mutant progerin—with a sticky substance that irreversibly attaches to any protein it comes within nanometers of. "It's like going fishing," explains Stewart. "You throw out a longline of your favorite protein, and then pull it in to see what proteins have hooked onto the line." The researchers compared the proteins that stuck to lamin-A with those that stuck to progerin.
Compared with lamin-A, progerin interacted significantly less with a protein also found in the nuclear lamina—lamina-associated polypeptide-α (LAP2α). The difference was significant enough to consider it a potential contender for preventing the accelerated biological aging. "I didn't think it would work, to be honest. We were looking for a needle in a haystack," says Dreesen.
As with telomerase, they introduced LAP2α to the progeric cells. To their amazement, the cells grew faster, had less DNA damage, and did not become unproductive prematurely. "We just couldn't believe it," says Dreesen. "We reproduced the experiment about seven times, but every time we did this, the cells grew better."
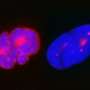
Using super-resolution microscopy, they further discovered that LAP2α was found in closer proximity to telomeres in normal cells than in progeric cells by almost 200 nanometers. This mislocalization of LAP2α might be responsible for the progerin-induced telomere defects.
The majority of telomeres in both types of cells were found within 250 nanometers of the nuclear lamina (see image). When taken as a whole, these findings set the scene for the pathology of progeria in which telomeres and the nuclear lamina play a prominent role.
Understanding how this process works could show scientists a way to prevent progeric cells from deteriorating and dying, says Dreesen. Ultimately, the researchers want to know how the changes observed in progeria apply to normal aging.
More information: Alexandre Chojnowski et al. Progerin reduces LAP2α-telomere association in Hutchinson-Gilford progeria, eLife (2015). DOI: 10.7554/eLife.07759
O. Dreesen et al. Lamin B1 fluctuations have differential effects on cellular proliferation and senescence, The Journal of Cell Biology (2013). DOI: 10.1083/jcb.201206121
Leslie C. Mounkes et al. A progeroid syndrome in mice is caused by defects in A-type lamins, Nature (2003). DOI: 10.1038/nature01631