Study describes the molecular cause of common cerebrovascular disease
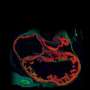
 in a mouse brain are visualized using microCT. Credit: Zinan Zhou, lab of Mark Kahn, Perelman School of Medicine, University of Pennsylvania")
Cerebral cavernous malformations (CCMs) are clusters of dilated, thin-walled blood vessels in the brain that can cause stroke and seizures, yet exactly how they form is somewhat of a mystery. Now, a team from the Perelman School of Medicine at the University of Pennsylvania has discovered the molecular mechanism that underlies this common cerebrovascular disease. They published their results this week online ahead of print in Nature.
CCM disease, which occurs in about one in 100 to 200 people, can present in two forms. One is sporadic, accounting for 80 percent of cases, and is most frequent in older individuals. The remaining 20 percent are familial, inherited cases. These patients present with a large number of lesions and more severe symptoms that arise in much younger individuals. CCM disease is a progressive disorder, and the standard of care remains surgical removal of the most dangerous lesions and symptom management.
When CCM was first identified as a genetic disease, mutations in one of two copies of three genes were identified in human patients. The proteins encoded by these genes were found to bind each other in a single complex, but this complex lacks intrinsic enzyme activity and how its loss results in vascular disease - and why this vascular disease arises so specifically in the brain - has remained unknown.
"Despite all we know about CCMs, how loss of the CCM complex causes disease remains controversial, with numerous downstream signaling pathways and processes proposed to play roles," said senior author Mark Kahn, MD, the Edward S. Cooper, MD/Norman Roosevelt and Elizabeth Meriwether McLure Professor in the department of Medicine. An important clue to disease pathogenesis was discovered while studying vertebrate heart development, when the Kahn lab found that CCM proteins in endothelial cells control the activity of the enzyme MEKK3 and a downstream gene expression program that is essential for the embryonic heart to properly develop.
For the Nature study, the Kahn lab investigated whether a conserved mechanism might also underlie the formation of CCM lesions in the postnatal brain. Using a neonatal mouse model of CCM disease, they found that expression of MEKK3 target genes, KLF2 and KLF4, was increased in the cells of newly formed CCM lesions. "From there we were able to demonstrate that partial, endothelial-specific loss of the genes for MEKK3, KLF2, or KLF4 completely rescues lesion formation and prevents the mice from dying due to CCM disease," Kahn said.
Consistent with these findings in mice discovered by co-first author and doctoral student Zinan Zhou, the team also demonstrated that expression of KLF2 and KLF4 is elevated in vessel-lining cells from human familial and sporadic CCM lesions. Studies by co-first author Alan Tang, an MD-PhD student, also in the Kahn lab, further revealed that a previously identified disease-causing human CCM2 mutation confers the disease by preventing the CCM protein complex from interacting with its signaling target MEKK3.
"Our studies conclusively identify that increased MEKK3 signaling and over-expression of KLF2 and KLF4 transcription factors are the molecular basis for CCM," Kahn said. Because the KLFs are MEKK3 targets, higher MEKK3 activity means higher KLF gene expression.
Analysis of single endothelial cells in lesions removed from the brains of patients with familial CCM revealed the presence of a "second hit," indicating that CCM lesions arise when endothelial cells become deficient in a CCM protein. Sporadic CCM has been thought to be the same disease as familial CCM because the brain lesions appear similar, but molecular and genetic data to support this idea have been sparse. Using antibodies that stain for KLF2 and KLF4, the Kahn lab also demonstrated that familial and sporadic CCM lesions arise due to the same molecular pathology, and therefore should be amenable to the same therapeutic strategies.
The pathology of CCM disease pertains to an adaptor protein complex, which bring proteins together in the right place at the right time to transmit a molecular signal. "Its job is as a go between," Kahn said. "It's a tool to make two other tools work, so it has been difficult to pinpoint its exact function and virtually impossible to design a therapeutic strategy to prevent CCM formation in patients."
The discovery that familial and sporadic CCMs both arise due to endothelial gain of MEKK3-KLF2/4 signaling may open the door for new therapies designed to block the activity of these proteins. In addition, the Kahn lab is actively investigating which KLF2/4 target genes may be responsible for creating CCM lesions. Parallel studies in the developing heart identified a critical role for ADAMTS enzyme, which degrade versican, a related CCM disease substance that is also highly expressed in the brain. "The present study demonstrates versican degradation surrounding the earliest CCM lesions, suggesting that this may also be a critical step in CCM pathogenesis and an important future therapeutic target," Kahn said.
More information: Cerebral cavernous malformations arise from endothelial gain of MEKK3-KLF2/4 signalling, Nature, DOI: 10.1038/nature17178