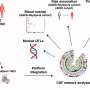
Brain's immune system triggers frontotemporal dementia
Frontotemporal dementia (FTD), the second most common cause of dementia in people under 65, may be triggered by a defect in immune cells called microglia that causes them to consume the brain's synaptic connections, according to new research led by UCSF scientists.
The new study – published April 21, 2016 in the journal Cell—adds to growing evidence that the brain's immune system is a driving force behind many neurodegenerative diseases, and suggests new approaches to diagnosing and treating patients with FTD, which currently affects as many as 22 out of 100,000 adults, with typical onset between the ages of 45 and 65.
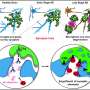
Microglia normally act as the brain's garbage collectors, disposing of foreign particles such as viruses or bacteria as well as sick and dying brain cells. In the developing brain, microglia also help refine the brain's circuitry by pruning back unneeded neural connections, which are marked for destruction with immune molecules called "complement proteins." Recent studies by Harvard neurobiologist Beth Stevens, PhD, and others have suggested that this process may go awry during adolescence in patients with schizophrenia, and as a side effect of aging in patients with Alzheimer's disease: in both cases an overabundance of complement protein appears to cause too many synapses to be tagged for destruction by the microglia.
"The brain's innate immune system is emerging as a common pathway behind many neurodegenerative disorders," said senior author Eric Huang, MD, PhD, a professor of pathology at UCSF and pathologist at the UCSF-affiliated San Francisco VA Medical Center. "This idea has been controversial, however, because in human patients, neurodegeneration is typically accompanied by some degree of inflammation, with lots of activated microglia, but it's hard to tell whether that is a driver of the degeneration or a consequence. You need careful experiments using animals models to dissect the cause-effect relationship."
In Mice, Gene Mutation Causes FTD-Like Symptoms
Working with colleagues at UCSF's Memory and Aging Center and departments of Neurology and Neurological Surgery, as well as the UCSF-affiliated Gladstone Institutes, Stanford University, and others, Huang and his team compared brain tissue from human FTD patients with familial mutations in the progranulin gene to the brains of mice with this gene deleted. In the mice, the defect caused age-related neurodegeneration and excessive grooming akin to the obsessive-compulsive disorder (OCD) symptoms seen in human FTD patients.
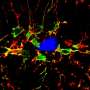
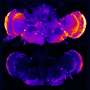
The researchers found that as the mice aged, the mutation caused a gradual breakdown of microglial cells' waste disposal systems, which led to excessive activation of these cells' aggressive immune functions, heightened production of complement proteins, and excessive synaptic pruning in the thalamus, a part of the brain that is highly relevant to human FTD.
Additional experiments on isolated microglia made it clear to the researchers that progranulin normally acts as a brake to prevent excessive microglia activation. Without it, it appeared that an unknown aspect of the normal aging process allowed microglia to spiral out of control.
However, the researchers showed that they could short-circuit this death spiral by deleting the gene for one of the major complement proteins produced by microglia, called C1qa. Mice with both the progranulin and the C1qa genes turned off lived considerably longer than those with intact C1qa, and didn't develop OCD-like behaviors. Their brains also showed a drastic reduction in the number of activated microglia and much better protection from synapse loss.
"We worked for more than two years to get to that result," Huang said, "But when we did, it was a really surreal experience. It was immediately clear that blocking complement protein might be a good therapeutic target for FTD patients with progranulin mutations."
Research Points to New Approaches for Dementia Patients
Huang and his team are now collaborating with a biotech company called Annexon to test therapies that block C1qa. However, these treatments are likely still a long way off. For one thing, treatments for neurodegeneration likely need to be taken early, before the brain damage is done, and there's still no way to reliably detect the disease before it's far too late.
That's why the researchers also investigated whether the elevated complement protein levels that would be predicted by their study show up in patients' cerebrospinal fluid (CSF)—the fluid collected by a spinal tap. If so, they could potentially serve as a biomarker to allow early detection of the disease.
Initially they were disappointed by the results: "We looked at CSF from normal and diseased brains side by side, and they looked about the same," Huang said. "But then we tried separating the FTD patients based on how severe their dementia had been when the CSF samples were taken, and the result was really spectacular. Clearly, as patients' mental status declined, the level of complement protein in their CSF increased."
Huang says he hopes future research will allow physicians to use this signal to enable earlier diagnosis and to test the effectiveness of potential treatments. If doctors give an anti-complement drug and the levels of complement protein in the CSF go down, he said, that would be a good sign that the immediate danger to the brain has been relieved.
More information: Cell, dx.doi.org/10.1016/j.cell.2016.04.001