Defects in the body's cell disposal system may contribute to the most common form of lupus
A casual observation about size differences in mice has led to the discovery that defects in a process for digesting dead cells called LC3-associated phagocytosis (LAP) may lead to a lupus-like autoimmune disorder. St. Jude Children's Research Hospital scientists led the research, which appears as an advance online publication today in the scientific journal Nature.
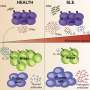
Lupus occurs when the immune system makes antibodies that target the patient's own tissue, causing widespread inflammation and life-threatening tissue and organ damage. The most common form of the disease, systemic lupus erythematosus (SLE), affects about 322,000 U.S. residents, primarily young women.
"We hope the findings offer a window into the cause of this devastating disease in some patients and an opportunity to improve treatment by developing novel approaches to prevent or reduce the inflammation and autoimmune response that characterize lupus," said co-corresponding author Douglas Green, Ph.D., St. Jude Department of Immunology chair.
LAP ensures that dead and dying cells are digested and disposed of properly after being eaten by immune scavenger cells called macrophages. In 2007, Green and his colleagues discovered LAP, which recruits components of another housekeeping system called autophagy to digest what the macrophages eat. Researchers subsequently showed that this helps to complete the job of removing a cell corpse.
First and co-corresponding author Jennifer Martinez, Ph.D., was a St. Jude postdoctoral fellow in Green's laboratory when she noticed that mice with a LAP defect were significantly smaller than related mice without the defect. Martinez is now with the National Institute of Environmental Health Sciences, Research Triangle Park, N.C.
The observation led researchers to the current discovery. The scientists found that LAP-deficient mice were not only smaller but also had increased levels of inflammation, autoimmune antibodies and other immune changes, including evidence of kidney damage, that are associated with SLE in humans. In contrast, defects in conventional autophagy did not lead to such changes.
When dying cells were injected into LAP-deficient mice, researchers found that macrophages responded normally but that, once engulfed, the dying cells were digested less efficiently than in mice without the defect. The LAP-deficient mice also had increased levels of immune molecules called cytokines that drive inflammation and a decrease in the anti-inflammatory cytokine interleukin 10 (IL-10).
Repeated exposure to dying cells accelerated signs of the lupus-like illness in LAP-deficient mice, including an increase in antibodies that attack normal tissue. That was not the case in mice in which LAP functioned normally. Defects in conventional autophagy also did not lead to problems digesting dead cells, increased production of inflammatory cytokines or lupus-like disease in mice.
The results were then confirmed by co-author Herbert Virgin, M.D., Ph.D., Department of Pathology and Immunology chair, Washington University School of Medicine, St. Louis, and his colleagues in mice from that institution.
LAP and autophagy are both systems that help cells isolate and dispose of threats. Some but not all of the same proteins are involved in both processes. For example, the protein NOX2 is required for LAP but not autophagy, and individuals who lack NOX2 often develop SLE.
"Defects in the clearance of dying cells have been implicated in the SLE disease process, and previous studies have suggested that the problem may stem from defects in autophagy," Green said. "In this study, we showed that LAP contributes to SLE in mice and likely accounts for the previously reported genetic association between autophagy and lupus."
Work has begun to identify LAP defects in humans with lupus and to explore a possible role for LAP in other inflammatory diseases.
More information: Jennifer Martinez et al, Noncanonical autophagy inhibits the autoinflammatory, lupus-like response to dying cells, Nature (2016). DOI: 10.1038/nature17950