Cancer metastasis: The unexpected perils of hypoxia
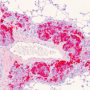
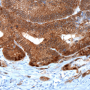
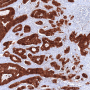
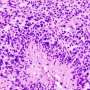
. Credit: Institute of Pathology, LMU Munich")
The low oxygen concentrations that prevail in many tumors enhance their propensity to metastasize to other tissues. Researchers at Ludwig-Maximilians-Universitaet (LMU) in Munich led by Professor Heiko Hermeking have now uncovered the molecular mechanism that links the two phenomena.
Many actively growing tumors are poorly supplied with blood, which limits the concentration of oxygen available within the tumor, a condition known as hypoxia. Tumor hypoxia has several significant consequences: The relative lack of oxygen partly accounts for the fact that such solid tumors are comparatively resistant to radiation and chemotherapy, and it also promotes the formation of satellite tumors in other tissues. Now researchers led by LMU professor Heiko Hermeking have dissected the mechanisms responsible for the association between tumor hypoxia and metastasis. Their results reveal how hypoxia leads to inhibition of the synthesis of a short RNA molecule that normally suppresses tumorigenesis. The new findings appear in the journal Gastroenterology.
Cancers can only develop if the biochemical circuits that control cell proliferation and behavior are inactivated. These safe-guarding mechanisms are mediated by so-called tumor-suppressor proteins, one of which is known as p53. The gene encoding p53 is inactivated in more than half of all tumors. In previous studies Hermeking had shown that p53 induces the transcription of a short RNA - referred to as microRNA-34a (miR-34a) - which plays a central role in tumor suppression. "We observed that, in patients with colon carcinomas, the gene for miR-34a is very frequently inactivated in metastasizing tumors, which are often characterized by relative oxygen deficiency," Hermeking says.
He and his colleagues have now shown that, in tumor cells in which the p53 function is compromised, complete loss of miR-34a expression is a direct consequence of hypoxia. In response to low oxygen levels, the tumor cells trigger the synthesis of hypoxia-inducible factor 1a (HIF1a), a protein that directly represses the transcription of miR-34a. Furthermore, this down-regulation of the microRNA is a prerequisite for hypoxia-induced epithelial-to-mesenchymal transition (EMT). In this process, HIF1a activates a genetic program that results in the transformation of non-invasive cells (which grow in a regulated fashion in epithelial sheets) into invasive, migratory cells that can seed new tumors elsewhere. EMTs play important roles in embryonic development and organogenesis, as well as in wound healing, but must be tightly regulated.
Among the proteins involved in orchestrating the EMT is PPP1R11. Hermeking's team found that especially high levels of this protein are synthesized in the cells that form the leading edge of an invasive tumor - where the oxygen concentration is expected to be particularly low. In normal cells, on the other hand, the production of PPP1R11 is repressed directly by miR-34a and thus indirectly by the tumor suppressor p53. This ensures that cells divide in a coordinated manner and prevents formation of metastases. "The regulatory antagonism is presumably the reason why metastasizing tumor cells are selected for loss of the p53 gene, which is required for the expression of miR-34a," Hermeking explains.
The new findings also have therapeutic implications, for they suggest that metastasizing colon tumors could perhaps be treated with drugs that inhibit the functions of proteins that promote the EMT. Such inhibitors would also be expected to permit reactivation of the expression of miR-34a. "In fact, molecules that act as functional substitutes for miR-34a are now being tested in clinical trials," says Hermeking. This miRNA is of particular interest as a drug target, because it is involved in the control of many regulatory processes. For instance, reintroduction of miR-34a into tumor cells also activates the patient's immune system to attack the tumor. Hermeking's results suggest that hypoxic tumors might be especially susceptible to this approach.
More information: Huihui Li et al, Antagonistic Effects of p53 and HIF1A on microRNA-34a Regulation of PPP1R11 and STAT3 and Hypoxia-induced Epithelial to Mesenchymal Transition in Colorectal Cancer Cells, Gastroenterology (2017). DOI: 10.1053/j.gastro.2017.04.017