Researchers discover fundamental pathology behind ALS

A team led by scientists at St. Jude Children's Research Hospital and Mayo Clinic has identified a basic biological mechanism that kills neurons in amyotrophic lateral sclerosis (ALS) and in a related genetic disorder, frontotemporal dementia (FTD), found in some ALS patients. ALS is popularly known as Lou Gehrig's disease.
The researchers were led by J. Paul Taylor, M.D., Ph.D., chair of the St. Jude Cell and Molecular Biology Department and a Howard Hughes Medical Institute investigator; and Rosa Rademakers, Ph.D., of the Mayo Clinic in Jacksonville, Florida. The findings appear today in the journal Neuron.
The disease-causing mutation identified is the first of its kind, Taylor said. Unlike in other genetic diseases, the mutation does not cripple an enzyme in a biological regulatory pathway. Rather, the mutation produces an abnormal version of a protein involved in a process called phase separation in cells.
Phase separation is a mechanism by which proteins assemble into organized assemblies, called membrane-less organelles, necessary for orderly cell functions. The researchers found that the ALS/FTD mutation produces an abnormal version of a protein called TIA1 that is a building block of such organelles. As a result, in ALS, the proteins within the organelles accumulate and kill neurons that control muscles. In FTD, the accumulation kills neurons in the brain. The researchers noted that abnormal phase separation may also underlie Alzheimer's disease.
There is currently no effective treatment for ALS/FTD. However, the researchers believe their finding offers a promising pathway for developing treatments to restore neurons' ability to disassemble the organelles when their cellular purpose has ended.
The TIA1 mutation was discovered when the scientists analyzed the genomes of a family affected with ALS/FTD. Tracing the effect of the mutation on TIA1 structure, the researchers found that it altered the properties of a highly mobile "tail" of the protein. This tail region governs the protein's ability to assemble with other TIA1 proteins. Taylor and his colleagues previously identified such unstructured protein regions, called prion-like domains, as the building blocks of cellular assemblies and as hotspots for disease-causing mutations.
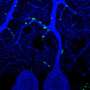
In further studies, the researchers found that TIA1 mutations occurred frequently in ALS patients. The scientists also found that people carrying the mutation had the disease. When the investigators analyzed brain tissue from deceased ALS patients with the mutations, the scientists detected a buildup of TIA1-containing organelles called stress granules in the neurons. Such granules form when the cell experiences such stresses as heat, chemical exposure and aging. To survive, the cell sequesters in the granules' genetic material that codes for cell proteins not necessary for survival-critical processes.
The granules also contained a protein called TDP-43, another building block of stress granules, whose abnormality has been implicated in causing ALS. In test tube studies and experiments with cells, the researchers found that the TIA1 mutation causes the protein to become more "sticky," delaying the normal disassembly of stress granules, trapping TDP-43.
"This paper provides the first 'smoking gun,' showing that the disease-causing mutation changes the phase transition behavior of proteins," Taylor said. "And the change in the phase transition behavior changes the biology of the cell."
More broadly, he said, "These findings are part of an emerging theme that there is a whole spectrum of diseases that includes ALS, and some forms of dementia and myopathy, that are caused by disturbance in the behavior of these structures that perturbs cellular organization."
The findings offer a highly promising pathway to the first effective treatments for ALS/FTD, Taylor said. Current drugs, which are only minimally effective, seek to improve the function of already damaged neurons. However, the new findings suggest the possibility of treatments that would prevent neuronal damage by restoring the healthy balance of phase separation in the cells of people with ALS/FTD mutations.
"We know that these material properties are under tight regulation, so perhaps we don't have to target the disease-causing mutation itself," Taylor said. "Perhaps we can restore balance by targeting any of a large number of regulatory molecules in the cell. There are already therapeutic approaches in laboratory testing that seek to do just that."
In further studies, Taylor and his colleagues will seek to understand the basic process of phase transition. They will also map the regulatory machinery for stress granules, to seek potential therapeutic targets. He also noted that the same basic pathology of phase transition may also underlie other neurodegenerative diseases, including Alzheimer's disease, and he is aiding researchers in applying the same research approach as in ALS/FTD to Alzheimer's.
More information: Neuron (2017). DOI: 10.1016/j.neuron.2017.07.025