New digestive / liver disease gene identified by international team
In a study published this month in Hepatology, a multinational team of researchers describes a newly identified cause of congenital diarrhea and liver disease in children.
The rare disorder is characterized by significant diarrhea beginning soon after birth, low serum levels of fat-soluble vitamins and evidence of liver disease. Despite continued symptoms, with medical support, the children grow and develop normally, at least to the age of 12.
Researchers from Emory University School of Medicine and Children's Healthcare of Atlanta , working with colleagues from Makassed Hospital, Al-Quds University and Hadassah Medical Center, Hebrew University of Jerusalem studied a family with two children from the Palestinian territories who suffer from the disorder.
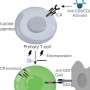
The team found that both children had inherited a mutation in a gene responsible for the transport of bile acids, which facilitate the digestion and absorption of dietary fats and fat-soluble vitamins. Although mutations had been identified in other genes important for the recycling of bile acids, this is the first report in humans of disease-associated defects in this gene, called Organic Solute Transporter-beta (SLC51B).
Almost 20 years ago, pediatric GI & hepatology researcher Paul Dawson, PhD, and colleagues identified mutations in another bile acid transporter gene (ASBT; SLC10A2) that caused congenital bile acid diarrhea.
"Even at that time, we knew that there were patients with similar symptoms that did not carry mutations in ASBT. But the genetic cause remained a mystery." Dawson says. "What's distinctive about this report is that these patients also have features of liver disease, which was not observed in previously described congenital bile acid diarrhea patients."
OST-beta, which works together with its partner protein Organic Solute Transporter-alpha, was the last of the major bile acid transporters to be discovered. Much less is known about its function and relationship to human disease.
"The effects of this mutation on the liver of these patients was a surprise, particularly because liver symptoms were not observed when mutations in OST-alpha were tested in mice," Dawson says.
Disease or syndrome-causing mutations had been discovered in the other major bile acid transporter genes, and the patients described in the current paper help close the loop. Moreover, the discovery will facilitate genetic identification of other patients and help guide their treatment.
"It was really gratifying to go from the discovery of the mechanism to the disease in patients," Dawson says.
Medications called bile acid sequestrants could help, along with fat-soluble vitamin supplementation and certain dietary changes and supplements. Since the signs of liver stress in these children appear to be milder than in other forms of childhood liver disease, the more aggressive clinical approaches used for those diseases may not be necessary. However, it is likely that there will be a spectrum of intestinal and liver abnormalities found in children who have OST-beta deficiency as this information is applied to help diagnose children with unknown and select GI and liver symptoms.
Mutaz Sultan, MD, a pediatric hepatologist at Makassed Hospital, identified the children and had ruled out other causes such as cystic fibrosis and ASBT deficiency that could account for their symptoms. Sultan and Bassam Abu Libdeh, MD, Medical Director and Chief of Pediatrics at Makassed Hospital, worked with Professor Orly Elpeleg, Head of the Department of Genetic and Metabolic at Hadassah Medical Center to perform whole exome sequencing, and with Dawson and colleagues at the Emory University School of Medicine to test the effects of the mutation in cells.
More information: onlinelibrary.wiley.com/doi/10 … 2/hep.29516/abstract