Research advance may prevent a form of hereditary hearing loss

A research advance co-led by Case Western Reserve University School of Medicine's Kumar Alagramam, PhD, may stop the progression of hearing loss and lead to significant preservation of hearing in people with Usher syndrome type III, a form of hereditary hearing loss linked to defects in the sensory "hair" cells in the inner ear. USH3 is caused by a mutation in the clarin-1 gene.
The onset of sensory deficits in USH3, which also can result in loss of vision, is generally noticeable in childhood and is primarily found in people of Finnish background and Ashkenazi Jewish descent.
In a new study published in Scientific Reports, Alagramam, who is an associate professor of otolaryngology, genetics and genomic sciences, and neurosciences at CWRU School of Medicine, and Lawrence Lustig, MD, chair of the Department of Otolaryngology—Head & Neck Surgery at Columbia University Medical Center, found that loss of hearing can be curtailed in a mouse model for hearing loss in USH3 through gene therapy. In gene therapy, normal genes are transplanted into cells in place of malfunctioning or missing ones to repair genetic disorders.
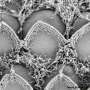
The clarin-1 gene provides information for making CLRN1, a protein found in hair cells, which help convey sound signals to the brain. "I think of hair cells as neatly arranged mini-microphones in the inner ear that are sensitive to different frequencies of sound entering the ear," said Alagramam. Previous work from the Alagramam lab showed that clarin-1 is essential for maintaining the structural integrity of the hair cells.
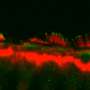
"There are several 'firsts' in our paper," said Alagramam. "This is the first time researchers have been able to successfully mimic in any animal the progressive hearing loss observed in USH3 patients with a disabling mutation in the clarin-1 gene." Previous research showed that there was already deterioration in the hair cells of mice with USH3 two or three days after birth, which explained why conducting gene therapy that early, even before the ear had fully developed, was already a lost cause. This precluded treating the cells before the onset of symptoms.
Seeking to address this problem, the authors surmised that inducing gradual hearing loss in mice would parallel the progression of hearing loss in humans with USH3, paving the way to investigate therapies for eventual human benefit. As reported in the new study, the researchers were able to postpone the onset of hearing loss and associated hair cell degeneration by about a month, providing enough time to inject normal copies of the Clarin-1 gene into the ear before the onset of hearing loss to see if the treatment was effective.
Secondly, this is the first research to demonstrate that the loss of hair cells and hearing can be curtailed in USH3 mice by clarin-1 gene therapy. Normal mice can hear 30 decibels, just above a human whisper. But by the age of 80-90 days, untreated USH3 mice in the study couldn't hear 100 decibels - akin to standing next to a running snowmobile or in a busy woodshop and not hearing anything. "We found that gene therapy blocked progressive hearing loss and improved hearing in treated mice by nearly four orders of magnitude compared to untreated sibling controls," said Alagramam. "The treated mice could hear at 45 decibels until about five months of age, when we concluded our study. This is significant preservation of hearing and could dramatically change the lives of hearing-impaired children and adults since, while the age of USH3 onset in people is typically three to ten years of age, hearing loss in USH3 can surface as late as age 30."
In a third "first," Alagramam and his colleagues revealed the potential importance of untranslated regions of genes in gene therapy. They showed that both the coded and the untranslated regions of the Clarin-1 gene are critical for effective gene therapy. Coded regions are portions of the gene that are translated into amino acids. Untranslated regions, once thought of as evolutionarily useless junk remnants, are now known to be vital for many of the regulatory aspects of genes.
"This is an important finding that will help future researchers, and eventually physicians, to maximize the effectiveness of gene therapy for curtailing sensory deficits associated with USH3 syndrome and, perhaps, in other disorders linked to defects in single genes," said Alagramam. "Our findings, combined with undertaking detailed family histories and, when appropriate, early genetic diagnosis, could eventually allow clinicians to replace the defective Clarin-1 gene before patients start to lose their hearing. While there is much work to be done before our approach can be tested in humans, it is reasonable to project that preservation of hearing, and preventing deafness, in humans with USH3 syndrome could become a reality within the next ten years."
More information: Ruishuang Geng et al, Modeling and Preventing Progressive Hearing Loss in Usher Syndrome III, Scientific Reports (2017). DOI: 10.1038/s41598-017-13620-9