A new test to measure the effectiveness of CF drugs
UNC School of Medicine researchers have developed a new laboratory model of the infection- and inflammation-plagued airways of cystic fibrosis (CF) patients. The model, described in the American Journal of Respiratory and Critical Care Medicine, includes primary bronchial epithelial cells from CF patients as well as infectious/inflammatory factors normally found in the CF airways. Pharmaceutical companies are now using the new model to test existing and potential CF therapies.
In their study, the UNC scientists showed how the model can be used to measure and compare the responses of CF and normal airway cells to CF-related infectious/inflammatory factors.
"This model is important because it contains the mix of infectious/inflammatory factors in the airways of CF patients," said principal investigator Carla M. P. Ribeiro, PhD, associate professor in the department of medicine and member of the UNC Marsico Lung Institute. "Understanding how airway cells respond to that environment gives us a better way to assess pre-clinically the likely effectiveness of novel CF therapies."
CF afflicts about 30,000 people in the United States, and is caused by inherited mutations in both copies of the gene that encodes the protein CFTR (cystic fibrosis transmembrane conductance regulator). In airway-lining cells, the protein controls the flow of chloride and water molecules between these cells and the thin layer of watery mucus that normally floats above them. That layer of mucus is supposed to carry inhaled bacteria and viruses out of the airways. But in CF, mucus is thick, sticky, and immobile, giving inhaled pathogens an environment in which they can thrive. The resulting chronic infections cause progressive lung damage, leading to respiratory failure. Treatments for CF include antibiotics, anti-inflammatory and mucus-thinning drugs, and even lung transplants, but most patients die before age 50.
Scientists have developed useful models of CF by engineering mice and other lab animals to have CFTR gene defects. However, to screen libraries of potential drug compounds to find any that work against CF, scientists typically need lab-dish (in vitro) models. The new in vitro model developed by Ribeiro and colleagues uses bronchial epithelial cells and mucopurulent materials from lungs that were removed from CF patients during lung transplants. The cells are exposed to a solution derived from the CF mucopurulent material, which contains bacterial factors and inflammatory factors secreted by airway epithelia and inflammatory cells that accumulate in CF lungs with frequent infections.
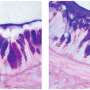
UNC research associate Lubna Abdullah, PhD, the first author of the study and Marsico member, found that long-term exposure to the infectious/inflammatory factors causes both CF and normal airway cells to boost their production of proteins known as mucins, the key gel-forming components of mucus.
Some previous research had suggested that CF airway cells, compared to healthy airway cells, overproduce or even underproduce mucins. However, Ribeiro said, "Our results show that the CF mucin response to infectious/inflammatory factors is really the same as that of normal airway cells."
The big difference in the cells' responses has to do with fluid secretion: The normal cells secrete more fluid to keep the increased mucus thin and mobile, whereas the CF cells secrete hardly any fluid.
"We showed unequivocally with this model that the lack of CFTR function in the CF airway cells leads directly to the dehydrated mucus seen in the disease," Ribeiro said.
She is now working with pharma companies to use the model to test prospective CF drugs. "This model has been very useful so far in our research, and I hope it will soon be useful more broadly for the CF research community," she said.