Biological engineers discover why a promising drug failed in clinical trials

Pharmaceutical companies once considered a protein called p38 a very attractive target for treating rheumatoid arthritis. Arthritis patients usually have elevated activity of this inflammation-producing protein, and in lab studies p38 inhibitors appeared to soothe inflammation. However, these drugs failed in several clinical trials.
A new study from MIT sheds light on just why these drugs did not work for arthritis. By untangling the complex interactions between different cell pathways involved in inflammation, the researchers discovered that shutting off p38 triggers other inflammatory pathways.
The findings demonstrate the importance of studying a potential drug's impact on complex cellular systems, says Doug Lauffenburger, head of MIT's Department of Biological Engineering and the senior author of the study. It's also important to do these studies under environmental conditions that match those found in diseased tissue, he adds.
"You've got to make sure you understand the complexity of the intracellular networks, and beyond that, you need to think about the environment you put the cells in," Lauffenburger says. "It's easy to get different results in different contexts, so you need to study them under many different conditions."
Former MIT postdoc Doug Jones is the lead author of the paper, which appears in the March 6 issue of Science Signaling.
A promising target
Rheumatoid arthritis, which afflicts more than 1 million Americans, is an autoimmune disorder that produces swollen and painful joints, primarily affecting the wrists and hands. This pain results from inflammation in the lining of the joints. Cells called synovial fibroblasts, which typically provide structural support for the joint lining, promote the inflammation and swelling in arthritic conditions.
Several years ago, scientists seeking new treatments for arthritis discovered that synovial fibroblasts from arthritis patients had very high levels of p38, and many pharmaceutical companies began working on p38 inhibitors. "The activity of this pathway was so strong that people tended to think that it was the best one to inhibit," Lauffenburger says.
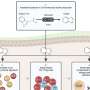
Despite their promise, p38 inhibitors failed in phase II clinical trials run by at least eight pharmaceutical companies. One of those companies, Boehringer Ingelheim, asked Lauffenburger to help them figure out why. Lauffenburger's lab focuses on systems biology, a field that involves measuring the interactions of many cell components and then performing computational modeling of those measurements to predict cell behavior.
The researchers' analysis revealed that the inflammatory pathway controlled by p38 interacts with several other pathways that can cause inflammation. These pathways, known collectively as stress pathways, produce inflammatory cytokines in response to events such as infection or injury.
The MIT team found that when p38 is extremely elevated, it suppresses the activity of these other inflammatory pathways. Therefore, when it gets turned off, the brake on the other pathways is released. Under these circumstances, inflammation remains high—the difference is that now it is controlled by other stress pathways.
"This is an insightful paper on redundancy in signaling and the need to understand compensatory mechanisms before spending billions on drug development. In that sense, it is a far more important insight than 'just' p38 inhibitors, and it makes clear again that animal efficacy models have severe limitations as tools to predict human efficacy," says David De Graaf, CEO and president of Syntimmune, who was not involved in the research. "This paper outlines one very thoughtful and generic approach to answer complex questions about efficacy in ex vivo human model systems."
Environment matters
Why was the MIT team able to see this phenomenon when others had not? Lauffenburger says one key is the environment in which the synovial fibroblast cells were studied.
Normally, cells studied in the lab are grown in a culture medium that offers them nutrients and molecules called growth factors, which keep the cells alive and proliferating. However, the MIT team found that under these conditions, a pro-growth pathway called MEK actually keeps p38 levels lower than in cells under stress. Because p38 is not as high, it doesn't inhibit the other stress pathways as strongly, so when the cells are exposed to p38 inhibitors, the other pathways don't soar into action and overall inflammation goes down.
"It looks like p38 inhibitors work well, if cells are in these growth factor environments," Lauffenburger says.
However, the MIT team found that synovial fluid from arthritis patients is not a pro-growth environment but is full of inflammatory cytokines. They then decided to expose synovial fibroblasts taken from patients with arthritis and from healthy individuals to this inflammatory environment. In both healthy and diseased cells, p38 levels skyrocketed, producing more inflammation and shutting off other stress pathways.
One question still to be answered is whether p38 inhibitors could work against other diseases such as cancer, in which the cells targeted would likely be in a pro-growth environment. They are also being considered as potential treatments for other inflammatory diseases such as multiple sclerosis and Alzheimer's. Lauffenburger says that their success will likely depend on what kind of environment the affected cells are in.
"A p38 inhibitor could work; you just have to know what the context is that the target cells are in. If you have the same kind of inflammatory cytokines there, then you might encounter the same problem" seen in arthritis, he says.
It's also possible that p38 inhibitors could work against arthritis or other drugs if given along with drugs that shut off other stress pathways, but more research would be needed to investigate that possibility, Lauffenburger says.
This story is republished courtesy of MIT News (web.mit.edu/newsoffice/), a popular site that covers news about MIT research, innovation and teaching.