Artificial gene defect reveals target to fight genetic disease

Fanconi anemia (FA) is caused by defective genes for DNA-repair leading to bone marrow failure, developmental abnormalities and increased cancer risk. Using genome-wide genetic approaches, researchers at CeMM systematically screened for the loss of an additional gene that could rescue the disease—and found it. The corresponding protein turned out to be a potential target that could be therapeutically exploited for FA. The study was published in Nature Communications.
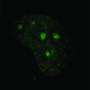
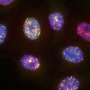
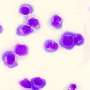
DNA repair is essential for a healthy organism. Every day, tens of thousands of DNA strands are damaged. Hence, it is not surprising that a broad variety of repair mechanisms developed in the course of evolution enables cells to quickly react and patch up the affected DNA strands. How important those repair mechanisms are becomes obvious when they fail—patients with FA are unable to repair DNA crosslinks, which, in most cases, eventually leads to cancer. So far, no curative therapy has been found for this disease.
Damaged DNA and its complex repair mechanisms comprise the research focus of the group of Joanna Loizou, principal investigator at CeMM Research Center for Molecular Medicine of the Austrian Academy of Sciences, and finding new molecular targets to fight FA is one of their goals. In their latest study, published in Nature Communications , the researchers aimed to find additional genes that genetically interact with the diseased FA genes and are essential for the manifestation of the disease, and thereby, if destroyed, restore the ability of the cell to repair DNA crosslinks. The research project was performed in collaboration with scientists from the University of Cambridge, from the Leiden University Medical Center, the University of California, the University of Toronto and the group of Jörg Menche at CeMM.
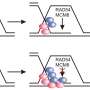
The scientists, with CeMM Ph.D. student Lydia Garcia-Robinson and former post doc of Loizou's lab Georgia Velimezi as shared first authors, deployed a novel genetic screen to search for synthetic viable interactions, using a genome-wide loss-of-function approach that uses insertional mutagenesis achieved via a gene-trap approach, on special lines of FA-defective cells that only possess one copy of each gene. With this method, they scored a bulls eye—the researchers found that an enzyme that removes ubiquitin, an important regulator of protein activity and half-life, causes FA gene deficiencies.

When the enzyme, called USP48, was artificially destroyed by CRISPR/Cas9, the FA-deficient cells were less sensitive to DNA-damaging compounds and showed an increased clearance of DNA damage. With further molecular analysis of the underlying processes, the researchers were able to show that the inactivation of USP48 in FA-deficient cells even restored a nearly error-free repair of the damaged DNA.
"Our results show that USP48 inactivation reduces chromosomal instability of FA-defective cells," Joanna Loizou explains. "This highlights a role for USP48 in controlling DNA repair and suggest it as a potential target that could be therapeutically exploited for Fanconi Anemia. To develop USP48 inhibitory molecules could be a new potential approach to alleviate the symptoms of FA patients."
More information: Georgia Velimezi et al, Map of synthetic rescue interactions for the Fanconi anemia DNA repair pathway identifies USP48, Nature Communications (2018). DOI: 10.1038/s41467-018-04649-z