Reproducing pediatric kidney disease from human iPS cells

Scientists in Japan have found a "skeleton key" for congenital kidney disease research. Using iPS cells generated from the skin cells of a patient with a nephrin mutation, Kumamoto University scientists have successfully developed kidney tissue that exhibits the early stages of congenital kidney disease. The protein nephrin is a constituent of the kidney filtration membrane, and abnormalities of this protein are commonly found in other types of kidney diseases.
The kidneys filter out and discharge waste products from the blood. During this process, proteins in the blood are prevented from leaking into the urine. The membrane responsible for this filtration is part of the glomerular podocyte, and nephrin is its main constituent. If there is a genetic mutation affecting the nephrin, a large amount of protein in the blood can leak into the urine, resulting in congenital nephrotic syndrome. The treatment of this syndrome is radical and difficult, and the absence of a technique to artificially reproduce a mutated filtration membrane has been a bottleneck in research progress.
In 2014, this Kumamoto University research group succeeded in the extremely difficult task of producing artificial kidney tissue in vitro from human iPS cells—a world first. And in 2016, they found that glomerular podocytes derived from iPS cells strongly express nephrin, that the human glomeruli readily connects with mouse blood vessels, and that induced podocytes continue to mature after transplantation into mice. With this knowledge, they applied their techniques to iPS cells derived from a single patient for their most recent project.
 with two methods, induction in vitro (left) or transplantation into mouse kidney tissue (right). The process took a total of about 1 month (13 days + 20 days). Credit: Prof. Ryuichi Nishinakamura")
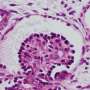
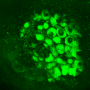
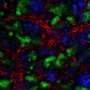
First, they established iPS cells from the skin cells of a patient with congenital nephrotic syndrome having a single nephrin mutation. When they induced kidney tissue from these iPS cells in vitro, they found that nephrin, which should exist on the surface of the glomerular podocyte, remained inside the cell, and that almost no filtration membrane precursor could develop. When podocyte maturation was performed by transplantation into mice, nephrin, which typically moves toward the blood vessel side of normal podocytes again remained inside the patient-derived cells. In other words, the initial pathology of this congenital kidney disease was reproduced by the iPS cells derived from the diseased patient.
Furthermore, when the nephrin mutation in patient-derived iPS cells were repaired and then induced into kidney tissue, the abnormalities were normalized. Put simply, the researchers found that this single mutation is the cause of the disease, and showed that it could possibly be treated by repairing the mutation.
, which normally exist on the sides and bottom of podocytes, are rarely detected in patients.Scale bar: 10 nanometers Credit: Prof. Ryuichi Nishinakamura")
"Because we can now reproduce the pathology of congenital nephrotic syndrome, it should be much easier to explore therapeutic drugs using this podocyte," said project leader Professor Ryuichi Nishinakamura. "Even when kidney disease develops in adults, it often begins with abnormal quantities of protein in the urine. It is believed that this is often caused by trouble in the nephrin protein of the filtration membrane. If a medication is discovered that controls nephrin protein, there is a strong possibility that it will be highly effective for kidney disease. What we have achieved here is a big step towards the development of drugs that act on podocytes and reduce proteinuria."
This research result was posted online in Stem Cell Reports on 30th Aug 2018.
More information: Shunsuke Tanigawa et al, Organoids from Nephrotic Disease-Derived iPSCs Identify Impaired NEPHRIN Localization and Slit Diaphragm Formation in Kidney Podocytes, Stem Cell Reports (2018). DOI: 10.1016/j.stemcr.2018.08.003