TGF-beta pathway protects against uterine cancer

Two new mouse models of uterine cancer shed light on how this disease—the most common gynecological cancer in the U.S. - happens. Led by researchers at Baylor College of Medicine, the team of scientists found that the Transforming Growth Factor beta (TGF-beta) signaling pathway in uterine cells protects against the disease by suppressing the overgrowth and transformation into cancer cells of the endometrium, the membrane lining the inside of the uterus.
The findings, which are published in two papers in the Proceedings of the National Academy of Sciences (PNAS) today, suggest potential new therapeutic strategies that could be beneficial to patients in the future.
"The TGF-beta signaling pathway regulates the development of many types of cancer, but it was not clear whether it also played a role in uterine cancer development," said corresponding author Dr. Martin M. Matzuk, director of the Center for Drug Discovery, Stuart A. Wallace Chair, Robert L. Moody, Sr. Chair and professor of pathology & immunology at Baylor College of Medicine.
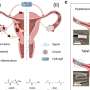
To explore the role the TGF-beta pathway plays in uterine cancer, the Matzuk lab developed two new mouse models of the disease. Each mouse model tested the effect of lacking specific proteins participating in the TGF-beta pathway; one model was studied in the absence of the protein TGF-beta receptor ALK5 and the other model tested the inactivation of both Smad2 and Smad3, proteins downstream of the receptor.
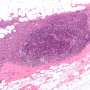
"The mice lacking ALK5 developed metastatic endometrial tumors with cervical and vaginal masses. The tumors were estrogen-dependent, meaning they required estrogen to grow. When the ovaries—the natural source of estrogen—were removed, both the tumors and metastases to the lungs regressed," Monsivais said. "These and other results indicate that it is important to maintain TGF-beta signaling via ALK5 to sustain the health and normal function of the uterus." This study is in this PNAS article.
The second mouse model was developed by Dr. Maya Kriseman, reproductive endocrinology and infertility fellow in the Department of Obstetrics and Gynecology at Baylor College of Medicine, and her colleagues. In this case, the researchers genetically modified mice to lack two other proteins, Smad2 and Smad3, which transmit the signal from the TGF-beta receptors to the nucleus. Then, they assessed the effect of lacking these proteins in tumor development.
"I found that disrupting these particular proteins, Smad2 and Smad3 in mice, caused a very aggressive hyperplasia or overgrowth of the endometrium that rapidly developed into cancer and death," Kriseman said. "These effects were dependent on estrogen, which also is a characteristic of the most common type of uterine cancer in humans. These and other findings give us a better idea as to why these mice develop aggressive uterine tumors and escape normal endometrial regulation." This study can be found in this PNAS paper.
"Taken together, the findings of Monsivais and Kriseman strongly suggest that the TGF-beta pathway is a tumor-suppressor mechanism in the uterus," said Matzuk, who also is a member of the Dan L Duncan Comprehensive Cancer Center at Baylor College of Medicine. "We anticipate that the new mouse models we have generated can be further used to uncover other processes that can lead to uterine cancer."
"Dissecting the complex cascade of biological events that are necessary for protecting the uterus from cancer and for sustaining normal fertility is critical. By revealing that the TGF-beta pathway plays a protective role against uterine cancer, this work has significantly advanced our understanding of the disease and opened novel avenues for future therapeutic strategies," said Dr. William E. Gibbons, professor of obstetrics and gynecology and director of reproductive endocrinology and infertility at Baylor College of Medicine.
More information: Maya Kriseman el al., "Uterine double-conditional inactivation of Smad2 and Smad3 in mice causes endometrial dysregulation, infertility, and uterine cancer," PNAS (2018). www.pnas.org/cgi/doi/10.1073/pnas.1806862116