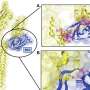
Cellular sickness linked to type 1 diabetes onset
 become senescent (red) before being attacked by the immune system. Right: A drug that eliminated senescent cells dramatically reduced T1 diabetes risk in mice. Credit: Bhushan lab / UCSF.")
A UC San Francisco study of human and mouse pancreatic tissue suggests a new origin story for type 1 (T1) diabetes. The findings flip current assumptions about the causes of the disease on their head and demonstrate a promising new preventative strategy that dramatically reduced disease risk in laboratory animals.
T1 diabetes is an autoimmune disorder that usually begins in childhood or adolescence, and has generally been attributed to the body's immune system mistakenly attacking the insulin-producing beta cells of the pancreas. Scientists still don't understand what triggers this autoimmune response, and so far, attempts to develop therapies to protect or restore beta cells have not been successful.
Now, in a study published February 21, 2019 in Cell Metabolism, UCSF Diabetes Center professor Anil Bhushan, Ph.D., and his team show that pancreatic beta cells themselves may play a much more active role in T1 diabetes than previously appreciated, opening the door to a totally new avenue for therapy.
Bhushan, who has long studied the biology of pancreatic beta cells, said that he has never been completely satisfied with the dominant model of the origins of type 1 diabetes: "Why does the immune system attack just those cells, while leaving neighboring cells of other types untouched?"
Dysfunctional Beta Cells Linked to Autoimmunity in Mice, Humans with T1 Diabetes
To understand whether beta cells might be contributing to early stages of T1 diabetes, a team led by Bhushan lab postdoctoral researchers Peter J. Thompson, Ph.D., and Ajit Shah, Ph.D., looked for changes in these cells during the early stages of disease development in so-called non-obese diabetic (NOD) mice, a commonly used animal model of the human disorder.
The researchers found that well before immune cells began to attack the pancreatic islets where beta cells reside, the beta cells began exhibiting signs of "secretory senescence," a type of cellular decline caused by DNA damage in which cells stop functioning properly and begin producing molecules that harm nearby cells and attract the attention of the immune system.
The new findings contrast sharply with the prevalent belief that T1 diabetes is caused by an overly aggressive immune system attacking healthy beta cells. The new data suggest instead that inherent problems with DNA repair in some beta cells triggers senescence, which patrolling immune cells first fail to recognize and clear out. As a result, these cells accumulate and spread so widely within the pancreas that by the time the immune system eventually recognizes the problem, it essentially has to raze the whole insulin-producing system, leading to the onset of diabetes.
"This is a paradigm shift for T1 diabetes therapy," Bhushan said. The main approach to date has been to dampen the immune system's attack on beta cells, but these data suggest the problem may not be an immune system gone awry. Instead, perhaps therapies should find a way to do the job the immune system is failing to do: clear the senescent cells early on."
To determine whether beta cell senescence plays a role in the onset of T1 diabetes in humans, the researchers studied pancreas tissue from deceased donors, sourced from the Network for Pancreatic Organ Donors with Diabetes, based at the University of Florida.
In line with their animal findings, the authors identified clear signs of DNA damage and secretory senescence in the beta cells of six donors with early-stage T1 diabetes, compared to six non-diabetic donors. The researchers also found signs of beta cell senescence in six donors without a diabetes diagnosis, but whose blood showed early signs of an immune reaction against beta cells, corroborating the idea that senescence is an early part of the chain of events leading to the disease.
"Seeing this data was an incredible moment," said Thompson. "Many results from these diabetic mouse lines have not panned out in humans, but the fact that we were seeing the very same markers of senescence in human pancreas tissue indicated that the same process is occurring in the human disease as well."
Eliminating Senescent Cells Prevents T1 Diabetes in Mice
To test whether eliminating senescent beta cells could help prevent T1 diabetes, Bhushan's group tested a drug called ABT-199 (Venetoclax), recently approved by the FDA as a second-line chemotherapy agent for a type of leukemia that also acts as a so-called senolytic—a drug that selectively eradicates senescent cells.
Remarkably, the researchers found that while 75 percent of control mice developed diabetes by 28 weeks of age, only 30 percent of mice who were given ABT-199 for two weeks prior to the onset of symptoms went on to develop the disease. The researchers showed that the drug had rapidly eliminated senescent beta cells in these mice, after which their immune systems (which were not directly affected by the treatment) left the remaining, healthy beta cells alone, avoiding the loss of insulin production that causes diabetes.
"These findings support the idea that senescent beta cells are like the bad apples that spoil the whole basket," Shah said. "Here we show that eliminating the bad apples can save the rest, which brings a new therapeutic avenue for treating patients with T1 diabetes."
Bhushan's group hopes these findings will lead to a therapy that could forestall the onset of T1 diabetes in young people at risk of developing the disease—which can currently be assessed via blood tests—and preserve remaining beta cell function in people with a recent T1 diabetes diagnoses. The animal experiments suggest that patients might be able to take such a drug periodically to clear out any senescent beta cells, and then perhaps be healthy for years.
"There is great excitement about the potential of senolytic drugs to treat all kinds of diseases of aging," Bhushan said. "Our work is among the first to suggest that clearing senescent cells can also be beneficial in pathological conditions not related to aging, such as type 1 diabetes."
More information: Cell Metabolism (2019). DOI: 10.1016/j.cmet.2019.01.021