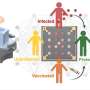
A link between mitochondrial damage and osteoporosis
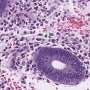
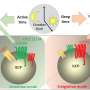
 were more likely to become osteoclasts—cells that break down bone—than control macrophages (left). Credit: Avadhani Lab")
Some risk factors for osteoporosis such as being older and female or having a family history of the condition cannot be avoided. But others can, like smoking cigarettes, consuming alcohol, taking certain medications, or being exposed to environmental pollutants. But until now researchers haven't gained a firm picture of how these exposures link up with bone loss.
A new study led by researchers from Penn's School of Veterinary Medicine reveals a mechanism by which these factors and osteoporosis may be linked. Damage to mitochondria—key cellular organelles and energy generators—leads to a surge in the creation of cells called osteoclasts, which are responsible for breaking down bone, the researchers report in FASEB Journal. They uncovered these effects in cells in culture as well as in an animal model.
"In a normal individual, the process of bone degradation and rebuilding proceeds in a very balanced way, but in some people they somehow produce a lot more osteoclasts, and this leads to bone loss and osteoporosis," said Narayan Avadhani, a biochemist at Penn Vet and senior author on the work. "We show in this paper that, when mitochondrial function is affected, it not only affects energy production but also triggers a type of stress signaling that induces the overproduction of osteoclasts."
Avadhani's lab has examined the effect of mitochondrial dysfunction on a variety of phenomena, including cancer and liver disease. Here, in collaboration with the lab of Mone Zaidi at Icahn School of Medicine at Mount Sinai, the scientists took a close look at how problems with mitochondria affected a type of immune cell known as macrophages.
Macrophages are a front line for the immune system, engulfing and digesting foreign invaders to the body. But macrophages can also diversify, transforming into osteoclasts when the circumstances are right.
To understand how mitochondrial damage could be linked to osteoporosis through the work of macrophages, the researchers induced damage to a key enzyme responsible for energy production in mitochondria, cytochrome oxidase C, in lab-grown mouse macrophages. Doing so led the macrophages to release a variety of signaling molecules associated with an inflammatory reaction and also seemed to encourage them to go down the path toward becoming osteoclasts.
Looking closely at what was going on, they observed an anomaly with a key molecule, RANK-L, that helps regulate the bone-rebuilding process and is released by bone-building cells as a means of inducing bone break-down. When mitochondria were damaged, they underwent stress signaling and transformed into osteoclasts at a much faster rate, even when RANK-L levels were low. These osteoclasts led to greater rates of bone resorption, or break down.
"In some respects, mitochondrial stress signaling may even be replacing RANK-L," says Avadhani. "That we don't know now, but we plan to look into that further."
The researchers confirmed their findings in a mouse model, showing that animals with a mutation that leads to dysfunctional mitochondria had increased production of osteoclasts. In addition, their macrophages had higher levels of phagocytosis, the process by which the immune cells engulf and "eat" invaders. The researchers believe this enhanced phagocytic ability could be responsible for some of the other physiological problems that can arise in people with defects in their mitochondria.
Because some of the same environmental risk factors that seem to promote osteoporosis, like smoking and some pharmaceuticals, can also impact mitochondrial function, the team posits that this stress signaling might be the pathway by which they are acting to affect bone health.
Future studies by the group, including Mount Sinai's Zaidi, a specialist in various aspects of osteoclast function and bone health, may look at how safeguarding mitochondrial function could protect against osteoporosis.
More information: Rajesh Angireddy et al, Cytochrome c oxidase dysfunction enhances phagocytic function and osteoclast formation in macrophages, The FASEB Journal (2019). DOI: 10.1096/fj.201900010RR