Tumor cells' drug addiction may be their downfall
. Credit: Matthew Sale, Babraham Institute")
Blocking the processes that drive cancer cell growth is at the heart of many new anti-cancer therapies. Unfortunately, after initial success, cancer cells are generally able to develop workarounds to reactivate the pathways that promote growth. Work by researchers at the Babraham Institute in partnership with the global biopharmaceutical company AstraZeneca shows that this workaround strategy proves fatal for the cancer cells once the treatment compound is withdrawn. The research is published today in the journal Nature Communications.
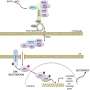
One of the major cell signalling pathways controlling cell growth and division is the RAS-BRAF-MEK-ERK pathway. Consisting of three enzymes working in a linear cascade, the pathway leads to the activation of ERK to promote cell division. Many cancers, including most melanomas and some colon cancers, arise due to mutations in the BRAF kinase, which results in an unprompted growth signal and inappropriate cell division.
Dr. Simon Cook and his team are specialists in the RAS-BRAF-MEK-ERK cell signalling pathway and through a long-standing partnership with collaborators at AstraZeneca had early access to a MEK inhibitor drug undergoing clinical testing to investigate how resistance to this drug develops.
"Tumours cells have shown themselves to be remarkably adaptable when treated with inhibitors of RAS-ERK signalling such as MEK or RAF inhibitors. Even in cases where these inhibitors are effective, invariably tumour cells adapt and acquire resistance to these drugs and their mechanisms of action," says Dr. Paul Smith, lead researcher at AstraZeneca. "Working with Simon Cook and his team at the Babraham Institute allowed us to apply their expert knowledge in cell signalling to better understand the mechanism of acquired resistance and excitingly, it has led to results that might change how MEK and other RAS-ERK pathway inhibitors, could be used for the treatment of cancer."
Simon Cook and his team used an AstraZeneca-developed MEK inhibitor called selumetinib (AZD6244/ARRY-142886) to study how resistance arises. After exposure of human cancer cell lines to selumetinib over several weeks, resistant cells arose through a process called gene amplification where the cells multiply their copies of their addicted oncogene, the mutated BRAF gene. This amplification of BRAF maintains growth signalling through ERK by overwhelming the presence of the drug working to shut down this pathway in much the same way as a tidal surge can overwhelm flood defences.
The researchers found that if they stopped treating the selumetinib resistant cells with the drug, their resistance to it was rapidly lost so that the tumour cells reverted back to being fully drug sensitive (by 5-10 weeks of growth in the absence of selumetinib depending on cell type).
The researchers were intrigued to find out why as this might tell us something important about how cell growth is controlled and might have relevance to the way new anti-cancer drugs are administered to patients.
They found that BRAF gene amplification becomes an impediment for cells in a drug-free environment because the cells are locked into abnormally high ERK activation. As a result of high ERK activity, a cellular ageing pathway is activated and the cells enter a permanent arrest of cell growth. Cells showing only modest levels of BRAF and ERK activity survive in this pool of cells and can divide but reacquire drug sensitivity so can be eliminated by a second wave of the drug.
"We know that cells need to maintain a level of activated ERK within a very specific range," explains Dr. Cook. "Rather than promoting more growth, too much ERK actually halts cell growth."
"In the presence of the inhibitor drug, the resistance mechanism sustains the cell's ERK level and cell growth," continues lead researcher Dr. Mathew Sale, a member of Simon Cook's group. "Once the inhibitor drug is gone however, the mechanism devised to maintain ERK signalling actually pushes the levels into the 'too high' zone—where the cell can no longer divide and often enters a phase of sustained growth inhibition and sometimes death."
In cell culture at least, this is a mechanism whereby the withdrawal of the anti-cancer drug can be used to eliminate resistant cells, clearing them from the population.
A previous study in laboratory mice has shown that intermittent dosing of similar drugs may elicit a more prolonged inhibition of tumour growth but the mechanism underlying this effect was not clear. This new study clearly defines the ERK 'sweet spot' as the determinant of reversibility, suggesting that in the case of cancers involving BRAF mutations, future clinical trials should consider intermittent dosing regimens to forestall the development of drug resistance.
More information: Matthew J. Sale et al, MEK1/2 inhibitor withdrawal reverses acquired resistance driven by BRAFV600E amplification whereas KRASG13D amplification promotes EMT-chemoresistance, Nature Communications (2019). DOI: 10.1038/s41467-019-09438-w