Taking out the protein garbage becomes more difficult as neurons age
 Time series of GFP-LC3B in the distal axon of a DRG neuron from a young adult mouse. Green and white arrowheads each follow one autophagosome biogenesis event. Retrograde is to the right. Scale bar, 2 μm. (B) Quantification of the rate of autophagic vesicle (AV) biogenesis (assayed by GFP-LC3B puncta formation per minute) in DRG neurons from young (one mo, light green), young adult (three mo, green), aged (16–17 mo, dark green), and advanced aged (24 mo, very dark green) mice (mean ± SEM; n ≥ 54 neurons from three biological replicates). Credit: Andrea Stavoe, Penn Medicine; eLife")
Cells dispose of harmful "trash" through autophagy, a normal and necessary process in which aggregated proteins and dysfunctional structures are handled. If any part of this fails, waste builds up inside cells, eventually killing them. According to a new study from the Perelman School of Medicine at the University of Pennsylvania, as cells age, their ability to shed harmful refuse declines. The findings suggest that the deterioration of autophagy in aged neurons—cells that never replicate and are as old as the bodies they inhabit—could be a risk factor for a suite of neurodegenerative diseases such as ALS (amyotrophic lateral sclerosis), Alzheimer's, and Parkinson's.
Using live-cell imaging of neurons from young and aged mice, Erika Holzbaur, Ph.D., a professor of Physiology, and first author Andrea Stavoe, Ph.D., a postdoctoral fellow in Holzbaur's lab, published their study this week in eLife. The importance of autophagy was recognized in 2016 with the Nobel Prize in Physiology or Medicine.
"The current thinking among scientists is that a decline in autophagy makes neurons more vulnerable to genetic or environmental risks," Holzbaur said. "What motivates our line of research is that most neurodegenerative diseases in which a deterioration of autophagy has been implicated, such as ALS, and Alzheimer's, Huntington's and Parkinson's diseases, are also disorders of aging,"
At the start of autophagy, a component within the cell, called an autophagosome, engulfs misfolded proteins or damaged structures to be degraded, essentially sequestering this waste in a biological trash bag. The autophagosome then fuses with a second cellular structure, called a lysosome, that contains the enzymes needed to breakdown the garbage, allowing the components to be recycled and reused. This elegant waste-removal stream is what keeps neurons healthy, but in its absence, neurons eventually die due to the buildup of unattended refuse.
"Think city streets during a sanitation workers strike," Stavoe said.
The team assessed rates of autophagy in mouse neurons during aging and identified a significant decrease in the number of autophagosomes produced, along with pronounced defects in the structure of autophagosomes produced by neurons from aged mice.
While early stages of autophagosome formation were unaffected, they found frequent stalling in their formation in aged mice, while the ones that did form were misshapen. These defects may allow the trash to accumulate at neuronal synapses. Stavoe notes that in other studies autophagosomes with misformed membranes have been observed in deceased human brain tissue from donors with neurodegenerative disease.
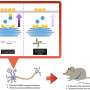
Importantly, turning on the protein WIPI2B in aged mice restores autophagosome formation in aged neurons, bringing the autophagy garbage-hauling process back online. This rescue is dependent on the level of activation of WIPI2B, providing insight into the biological regulation of autophagosome formation.
On the other hand, when researchers took WIPI2B out of young neurons, autophagosome formation stalled. "This stunning and complete rescue of autophagy using one protein suggests a novel therapeutic target for age-associated neurodegeneration," Stavoe said.
More information: Andrea KH Stavoe et al. Expression of WIPI2B counteracts age-related decline in autophagosome biogenesis in neurons, eLife (2019). DOI: 10.7554/eLife.44219