Protein aggregation: Protein assemblies relevant not only for neurodegenerative disease

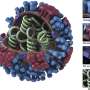
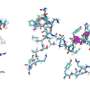
Amyloid fibrils play a crucial role in neurodegenerative illnesses. Scientists from Heinrich Heine University Düsseldorf (HHU) and Forschungszentrum Jülich have now been able to use cryo-electron microscopy (cryo-EM) to decode the spatial structure of the fibrils that are formed from PI3K SH3 domains—an important model system for research. Although the fibrils examined are not themselves connected with an illness, the findings made and methods developed could serve to understand diseases such as Alzheimer's and Parkinson's.
Proteins are central components of living material. These complex molecules made up of combinations of individual amino acids in some cases comprise thousands of individual atoms and have sophisticated three dimensional shapes. The term 'fold' is used to describe this structure. The fold of a protein determines its biological function.
Misfolding into non-natural structures and associated aggregation makes proteins not only useless but potentially toxic. The current view is that many neurodegenerative diseases are triggered by misfolded proteins. They form deposits in critical parts of the central nervous system. Initially, fibrillar structures, referred to as 'amyloid fibrils,' form. Larger deposits of such amyloid fibrils form the typical plaques that can be found in the brain tissue and can restrict, damage or kill nerve cells.
The PI3K SH3 domains are usually part of larger proteins, but can also exist alone in their correctly folded form. They play a major role in cellular communication. For many years, these domains have been used as model systems in order to examine protein folding and thus determine the causes of misfolding. This is because researchers have discovered that these domains can also form amyloid fibrils that do not differ from the fibrils typical for diseases and are just as poisonous to cells. In fact, all proteins can potentially form amyloid fibrils; healthy organisms must actively and constantly combat this process.
Many fundamental discoveries of amyloid fibrils that are directly applicable to disease-related proteins were made using this model system. "But what we didn't know until now was the precise three-dimensional structure of the fibrils from the PI3K SH3 domains," explains Prof. Dr. Gunnar Schröder, Professor of Computational Structural Biology at HHU as well as work group leader at Forschungszentrum Jülich.
"Now we can use cryo-EM to understand these structures fully," adds Prof. Dr. Alexander Büll, corresponding author alongside Schröder of the study published in Nature Communications. Büll was an Assistant Professor at HHU until early 2019 and is now a Full Professor in the Department of Biotechnology and Biomedicine at the Technical University of Denmark in Lyngby. Speaking about the significance of this determined structure, Prof. Schröder adds: "Now that we know the spatial structure, much of the earlier data from the last 20 years can be reinterpreted or interpreted more quantitatively."
"Cryo-electron microscopy is a wonderful tool for determining the three-dimensional structure of the fibrils," emphasises Christine Röder, first author of the study and a member of Prof. Schröder's work group in Jülich. The 2017 Nobel Prize in Chemistry was awarded for the development of this method, with which complex biomolecules that adopt their natural form only in an aqueous environment can be presented in atomic resolution. The samples dissolved in water—for example proteins—are plunge-frozen to very low temperatures and thus fixed in their natural structure. This makes it possible to examine them under an electron microscope in this state. However, this can't be achieved with a single image, but in a succession of several recordings that show the protein from different angles. Computers then put the many individual recordings together to form a three-dimensional image.
More information: Christine Röder et al, Atomic structure of PI3-kinase SH3 amyloid fibrils by cryo-electron microscopy, Nature Communications (2019). DOI: 10.1038/s41467-019-11320-8