Gene therapy for Duchenne muscular dystrophy safely preserves muscle function

A gene therapy being developed at Penn Medicine to treat Duchenne muscular dystrophy (DMD) successfully and safely stopped the severe muscle deterioration associated with the rare, genetic disease in both small and large animal models, according to a first-of-its-kind study from Penn Medicine researchers. The findings, published online today in Nature Medicine, puts the field within closer reach of a safe and effective gene therapy that uses a "substitute" protein without triggering immune responses known to hinder other therapeutic approaches.
Found mostly in boys, DMD is caused by mutations in a sex-linked gene that stop production of a muscle-building protein known as dystrophin. Without it, muscles progressively deteriorate and weaken starting at a very young age and only worsen from there. Most patients aren't able to walk by age 12 and die of heart or respiratory failure by the time they reach their 30s, though respirators have helped some live longer.
With their modified gene therapy approach, a multidisciplinary team from the Perelman School of Medicine at the University of Pennsylvania engineered adeno-associated virus (AAV) vectors to deliver a "substitute" protein for dystrophin in small and large animal DMD models to keep the muscles intact. The synthetic substitute, based on a naturally occurring protein called utrophin, proved to be an effective and safe alternative, as it protected muscle in mice and dogs with naturally occurring DMD-like mutations, including a large deletion that closely mirrors the large dystrophin deletions found in humans.
"For the first time, we've shown how a carefully constructed version of a dystrophin-related protein can safely prevent the breakdown of muscle and maintain its function over time in the most informative animal models. This discovery has important implications for gene therapy and how we work toward safe and effective treatments for muscular dystrophy," said senior author Hansell H. Stedman, MD, an associate professor of Surgery. "With these results, we have a strong rationale to move this forward into human clinical trials."
Restoring levels of dystrophin with gene therapy and other techniques have been under scrutiny due to the immune system's adverse reaction to additions it deems foreign. DMD patients have little to none of this protein, so their bodies can attack well-intentioned direct replacement proteins because they are seen as foreign. However, because dystrophin's distant cousin utrophin is expressed in other places in the body, it was thought that the immune system wouldn't view it as a threat.
The Penn team showed this to be true in rigorous, randomized mouse and dog studies. Delivering a single-dose treatment of synthetic utrophin with the AAV vector to newborn mice showed distribution of the protein throughout the body, no signs of toxicity, and complete suppression of all signs of DMD, compared to untreated mice. The mice also sustained expression of utrophin in skeletal and cardiac muscles throughout that time, and physical tests in the mice supported the sustained muscle function.
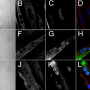
The team further investigated utrophin by administering it to dogs four to seven days of age in a randomized study. Six weeks after receiving a dose, the researchers observed a robust expression of utrophin and a four-fold increase in weight compared to previously reported weight loss and body-wide inflammation, suggesting induced "auto"-immunity in dogs who received human dystrophin. The Penn group also observed a significantly reduced level of muscle damage in the treated dogs.
In perhaps the most important canine study, researchers compared utrophin-treated limbs with dystrophin-treated limbs, and after four weeks, observed stark differences. Muscle biopsies revealed persistent expression of utrophin and suppression of ongoing muscle damage, but only sparse amounts of dystrophin in dying cells in the other limb. The immune responses also varied greatly between the limbs.
"Under the microscope it looked like a hand grenade had gone off in the limbs with dystrophin," Stedman said. "The experiments showed that the immune response to dystrophin was 100 to 1,000 times stronger than it was to utrophin."
Treated dogs also had nearly complete prevention of muscle degeneration and regeneration in their jaw closing (biting) muscles. These muscles, by virtue of their extreme power, are among the first to deteriorate in untreated dystrophic dogs.
This is the first large animal study to show utrophin's effectiveness as well as its non-immunogenic response. Taken together, the researchers said, these findings may refocus the field toward the use of a functionally optimized, safe utrophin-based gene therapy approach as the pathway to a potential cure for Duchenne muscular dystrophy.
To create Penn's synthetic version, a team of researchers first turned to evolutionary biology to better understand dystrophin's origin and how it was assembled, looking back and reconstructing genetic events during the earliest parts of Earth's history. Ongoing studies would reveal more about the protein's makeup, the strength of its rod structure, and its deletions, among other important characteristics, to inform future development.
"We have gained a lot of insight from that approach," Stedman said. "One of the things that we hope to do during clinical development is to leverage that insight to make ever better therapies, the strongest possible versions of 'nanotrophin'."
More information: Non-immunogenic utrophin gene therapy for the treatment of muscular dystrophy animal models, Nature Medicine (2019). DOI: 10.1038/s41591-019-0594-0 , nature.com/articles/s41591-019-0594-0