BIN1 deficit impairs brain cell communication, memory consolidation

Bridging integrator 1, known as BIN1, is the second most common risk factor for late-onset Alzheimer's disease, according to genome-wide studies of genetic variants. Yet, scientists know little about what this protein does in the brain.
Now a new preclinical study has discovered that a lack of BIN1 leads to a defect in the transmission of neurotransmitters that activate the brain cell communication allowing us to think, remember and behave. Led by Gopal Thinakaran, Ph.D., of the University of South Florida Health (USF Health) Morsani College of Medicine and colleagues at the University of Chicago, the study was published March 10 in Cell Reports.
Approximately 40% of people with Alzheimer's disease have one of three variations in the BIN1 gene—a glitch in a single DNA building block (nucleotide) that heightens their risk for the neurodegenerative disease, said the paper's senior author Dr. Thinakaran, a professor of molecular medicine at the USF Health Byrd Alzheimer's Center and associate dean for neuroscience research at the Morsani College of Medicine.
"Our findings that BIN1 localizes right at the point of presynaptic communication and may be precisely regulating neurotransmitter vesicle release brings us much closer to understanding how BIN1 could exert its function as a common risk factor for Alzheimer's disease," Dr. Thinakaran said. "We suspect it helps control how efficiently neurons communicate and may have a profound impact on memory consolidation—the process that transforms recent learned experiences into long-term memory."

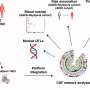
The research team created a mouse model in which the BIN1 gene was selectively inactivated, or knocked out, to characterize the protein's normal function in the brain. In particular, they used advanced cell and molecular biology techniques to investigate the role of BIN1 in regulating synapses associated with learning and memory.
To frame the study results, it helps to know that a healthy human brain contains tens of billions of brain cells (neurons) that process and transmit chemical messages (neurotransmitters) across a tiny gap between neurons called a synapse. In the Alzheimer's disease brain, this synaptic communication is destroyed, progressively killing neurons and ultimately causing a steep decline in memory as well as other signs of dementia. Individuals most susceptible to developing full-blown Alzheimer's in later life are those who lose the most synapses, Dr. Thinakaran said.
Among the Cell Reports study highlights:
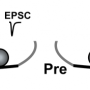
 combined with pre- and postsynaptic sites. White arrows at right indicate the overlap between BIN1 and synapsin, a protein involved in regulating neurotransmitter release at synapses. Credit: Gopal Thinakaran, USF Health Morsani College of Medicine, appeared first in Cell Reports (supplementary materials) doi.org/10.1016/j.celrep.2020.02.026")
- Loss of BIN1 expression in neurons leads to impaired spatial learning and memory. That is, the deficit alters how effectively information about surrounding environmental space is acquired, stored, organized and used. The BIN1 knockout mice had significantly more difficulty than controls in finding the hidden platform in a Morris water maze.
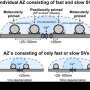
- Further analysis distinguished that BIN1 primarily locates on neurons that send neurotransmitters across the synapse (presynaptic sites) rather than residing on those neurons that receive the neurotransmitter messages (postsynaptic sites). Synaptic transmission in the hippocampus, a brain region associated primarily with memory, showed deterioration in the release of neurotransmitters from vesicles. Vesicles are bubble-like carriers that transfer neurotransmitters from presynaptic to postsynaptic neurons.
- The BIN1 deficiency was associated with reduced density of synapses and a decrease in the number of synaptic clusters in the knockout mice compared to controls.
- 3-D electron microscopy reconstruction of the synapses showed a significant accumulation of docked and reserve pools of synaptic vesicles in the BIN1 knockout mice. That indicates slower (less successful) release of neurotransmitters from their vesicles, the researchers suggest.
The study authors conclude that altogether their work highlights a non-redundant role for neuronal BIN1 in presynaptic regulation and "opens new paths for the future investigation of the precise role of BIN1 as a risk factor in Alzheimer's disease pathophysiology."
More information: Pierre De Rossi et al, Neuronal BIN1 Regulates Presynaptic Neurotransmitter Release and Memory Consolidation, Cell Reports (2020). DOI: 10.1016/j.celrep.2020.02.026