Researchers identify key mechanisms involved in pulmonary fibrosis development

Working alongside research groups from Heidelberg, researchers from Charité - Universitätsmedizin Berlin have elucidated the novel disease processes involved in the development of pulmonary fibrosis. They were able to show that the protein known as NEDD4-2 plays a key role in lung health and that loss of this crucial regulatory molecule has a significant impact on various mechanisms involved in the development of chronic progressive lung disease. These new insights make it easier to further investigate the precise mechanisms involved in the development and progression of pulmonary fibrosis. The researchers' findings, which have been published in Nature Communications, will enable researchers to develop new therapeutic approaches.
Pulmonary fibrosis is a serious lung disease which mainly affects older people; there are virtually no effective treatments. The disease is characterized by progressive tissue changes which lead to scarring of the lung. However, its causes are largely unknown, and the cellular mechanisms involved in its development remain poorly understood. The term 'mucociliary clearance' refers to a self-cleaning mechanism which relies on ciliated cells in the lung epithelium propelling inhaled pathogens and other particles trapped in mucus out of the airways. We know that changes in the epithelium are associated with the production of excess mucus. They are also associated with the impaired clearance of this mucus and its primary structural components (known as 'mucins'). NEDD4-2 is involved in the degradation of a range of other proteins which in turn are responsible for using these processes to regulate lung epithelial function. This means NEDD4-2 is a key protein with a central role in the pathogenesis of pulmonary fibrosis.
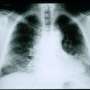
Working alongside colleagues from the German Center for Lung Research (DZL), Heidelberg University Hospital and the German Cancer Research Center, the team of researchers led by Prof. Dr. Marcus Mall (Director of Charité's Department of Pediatric Pulmonology, Immunology and Critical Medicine and Professor at the Berlin Institute of Health [BIH]) succeeded in developing a novel animal model of idiopathic pulmonary fibrosis (IPF). As NEDD4-2 is crucial for early development, the researchers only deleted the relevant encoding gene in lung epithelial cells once the animals had reached adulthood. The researchers examined the animals once they had reached a stage roughly comparable to the point at which the disease would be diagnosed in a human patient. Oxygen saturation measurements taken at that stage revealed a level of lung function impairment which is characteristic of the disease. Using tissue sections and CT imaging to examine the lungs, the researchers also found evidence of patchy scarring, a type of structural abnormality which is indicative of fibrosis. The researchers found further evidence of the significance of NEDD4-2 in the pathogenesis of IPF: lung tissue biopsy samples of patients with IPF contained significantly reduced levels of both transcripts and proteins. Using mass spectrometry, the researchers then performed what is known as 'protein profiling', an analysis of the complete set of proteins produced in the lungs. This revealed a high degree of overlap between the proteins found to be expressed differently in the lungs of patients with IPF and in the animals used in this study. "Our findings can help researchers to further investigate the pathogenesis and progression of this lung disease and develop new treatments. This model could prove useful for the preclinical testing of compounds with therapeutic potential, or to develop markers for the early detection of the disease," says Prof. Mall.
When studying the underlying disease mechanisms, the researchers discovered that reduced levels of NEDD4-2 in epithelial cells result in epithelial remodeling in the airways. Not only are the different cell types present in different proportions, the cells also produce increased amounts of certain mucins. When combined with changes in epithelial sodium transport and the resultant reduction in the volume of airway surface liquid, this will lead to impaired mucociliary clearance. Lack of NEDD4-2 also causes increased activity of the TGFβ signaling pathway, which promotes the formation of fibrosis. Summing up the findings of the study, the study's first author, Dr. Julia Dürr, says: "This means we were able to establish that a lack of NEDD4-2 is directly linked with mucociliary clearance dysfunction and the dysregulation of the TGFβ signaling pathway. According to current state of knowledge, both of these play a role in the pathogenesis of IPF."
Anti-fibrotic drugs have been used to treat pulmonary fibrosis for some years. While these drugs usually succeed in slowing the development of scarring, they cannot wholly replace lung transplants as a last resort treatment option. "Using our model and an already licensed anti-fibrotic agent, we were able to confirm that this type of treatment can slow disease progression but does not constitute a curative treatment." By way of explanation, Prof. Mall adds: "We hope that, by providing an improved preclinical testing model, we can help speed up the development of new therapeutic options." As a next step, the researchers plan to test predictive biomarkers which might aid in early diagnosis. They also plan to test the effectiveness of potential new drugs which could be used to treat pulmonary fibrosis.
More information: Duerr J et al. Conditional deletion of Nedd4-2 in lung epithelial cells causes progressive pulmonary fibrosis in adult mice. Nat Commun (2020), DOI: 10.1038/s41467-020-15743-6.