Your genes could determine whether the coronavirus puts you in the hospital
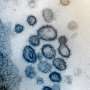
 bound to a piece of a virus (orange and blue) – in this case, influenza. Credit: Prot reimage via Wikimedia Commons, CC BY-SA")
When some people become infected with the coronavirus, they only develop mild or undetectable cases of COVID-19. Others suffer severe symptoms, fighting to breathe on a ventilator for weeks, if they survive at all.
Despite a concerted global scientific effort, doctors still lack a clear picture of why this is.
Could genetic differences explain the differences we see in symptoms and severity of COVID-19?
To test this, we used computer models to analyze known genetic variation within the human immune system. The results of our modeling suggest that there are in fact differences in people's DNA that could influence their ability to respond to a SARS-CoV-2 infection.
What we did
When a virus infects human cells, the body reacts by turning on what are essentially anti-virus alarm systems. These alarms identify viral invaders and tell the immune system to send cytotoxic T cells—a type of white blood cell—to destroy the infected cells and hopefully slow the infection.
But not all alarm systems are created equal. People have different versions of the same genes—called alleles—and some of these alleles are more sensitive to certain viruses or pathogens than others.
To test whether different alleles of this alarm system could explain some of the range in immune responses to SARS-CoV-2, we first retrieved a list of all the proteins that make up the coronavirus from an online database.
We then took that list and used existing computer algorithms to predict how well different versions of the anti-viral alarm system detected these coronavirus proteins.

Why it matters
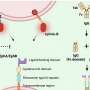
The part of the alarm system that we tested is called the human leukocyte antigen system, or HLA. Each person has multiple alleles of the genes that make up their HLA type. Each allele codes for a different HLA protein. These proteins are the sensors of the alarm system and find intruders by binding to various peptides—chains of amino acids that make up parts of the coronavirus—that are foreign to the body.
Once an HLA protein binds to a virus or piece of a virus, it transports the intruder to the cell surface. This "marks" the cell as infected and from there the immune system will kill the cell.
In general, the more peptides of a virus that a person's HLAs can detect, the stronger the immune response. Think of it like a more sensitive sensor of the alarm system.
The results of our modeling predict that some HLA types bind to a large number of the SARS-CoV-2 peptides while others bind to very few. That is to say, some sensors may be better tailored to SARS-CoV-2 than others. If true, the specific HLA alleles a person has would likely be a factor in how effective their immune response is to COVID-19.
Because our study only used a computer model to make these predictions, we decided to test the results using clinical information from the 2002-2004 SARS outbreak.
We found similarities in how effective alleles were at identifying SARS and SARS-CoV-2. If an HLA allele appeared to be bad at recognizing SARS-CoV-2, it was also bad at recognizing SARS. Our analysis predicted that one allele, called B46:01, is particularly bad with regards to both SARS-CoV-2 and SARS-CoV. Sure enough, previous studies showed that people with this allele tended to have more severe SARS infections and higher viral loads than people with other versions of the HLA gene.
Whats next?
Based on our study, we think variation in HLA genes is part of the explanation for the huge differences in infection severity in many COVID-19 patients. These differences in the HLA genes are probably not the only genetic factor that affects severity of COVID-19, but they may be a significant piece of the puzzle. It is important to further study how HLA types can clinically affect COVID-19 severity and to test these predictions using real cases. Understanding how variation in HLA types may affect the clinical course of COVID-19 could help identify individuals at higher risk from the disease.
To the best of our knowledge, this is the first study to evaluate the relationship between viral proteins across a wide range of HLA alleles. Currently, we know very little about the relationship between many other viruses and HLA type. In theory, we could repeat this analysis to better understand the genetic risks of many viruses that currently or could potentially infect humans.
This article is republished from The Conversation under a Creative Commons license. Read the original article.![]()
















{kind=link}
{kind=link}