Genetic doppelgaengers: Research provides insight into two neurological puzzles

An international team led by Emory scientists has gained insight into the pathological mechanisms behind two devastating neurodegenerative diseases. The scientists compared the most common inherited form of amyotrophic lateral sclerosis and frontotemporal dementia (ALS/FTD) with a rarer disease called spinocerebellar ataxia type 36 (SCA36).
Both of the diseases are caused by abnormally expanded and strikingly similar DNA repeats. However, ALS progresses quickly, typically killing patients within a year or two, while the disease progression of SCA36 proceeds more slowly over the course of decades. In ALS/FTD it appears that protein products can poison cells in the nervous system. Whether similar protein products exist in SCA36 was not known.
What Zachary McEachin, Ph.D. and Gary Bassell, Ph.D. from Emory's Department of Cell Biology, along with a team of collaborators at Emory, Mayo Florida, and internationally from Spain and Japan, discovered have provided a new paradigm for thinking about how aberrant protein species are formed. Despite the disparate clinical outcomes between these diseases, this research could broaden the avenue of research toward genetically targeted treatments for such related neurodegenerative diseases.
Their study, published Tuesday in Neuron, provides a guide to types of protein gobbledygook that build up in brain cells in both disorders, and which should be reduced if the new mode of treatment is working in clinical trials.
"We are thinking of these diseases as genetic doppelgängers," says McEachin, a postdoctoral fellow in Bassell's lab. "By that, I mean they are genetically similar, but the neurodegeneration progresses differently for each disease. We can use this research to understand each of the respective disorders much better—and hopefully help patients improve their quality of life down the road with better treatments."
An estimated 16,000 people in the United States have ALS, a progressive neurodegenerative disease that affects nerve cells in the brain and spinal cord. The most common inherited form of ALS/FTD occurs because there is an abnormally expanded repeat of six DNA "letters" stuck into a gene called c9orf72.
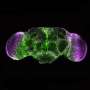
Some of the degeneration of motor neurons comes from the expanded repeat being made into proteins that repeat two amino acids over and over. It's like a printing press being forced to print entire pages of "GPGPGPGP", "GAGAGAGA"..." or "PRPRPRPR" In c9 ALS, those proteins are thought to build up inside brain cells and poison them. A striking finding is that in c9 ALS, there are others chimeric variants e.g. "GAGAGAGPGPGP" suggesting these toxic proteins are more complex than previously thought.
"A major goal in the field has been to identify and characterize the various types of pathological aggregates that accumulate in patient brains as 'bad actors' that cause neurodegeneration," Bassell says. "Here we identify fundamental differences in this process for these two neurological diseases with disparate clinical outcomes. I think this is a paradigm shift for how heterogenous this process is in ALS and points to possible therapeutic strategies that might mitigate multiple bad actors simultaneously."
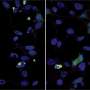
The two diseases (c9 form of ALS and SCA36) both have abnormally expanded 6-letter genetic repeats, but the letters that are being repeated are different. McEachin was able to show that the nonsensical protein products from the expanded repeats aggregate in c9 ALS patients' cells but do not in SCA36. He says it was surprising because the proteins were predicted to be similar in the two diseases.
"Studying the differences in the aberrant proteins gives insight into both diseases," McEachin says.
SCA36, one of several types of SCA, is a condition characterized by progressive problems with movement that typically begins in mid-adulthood. They also develop hearing loss and muscle twitches over time, including losing the ability to move their tongue. Their legs, forearms and hands atrophy as the condition worsens.
The mutation for SCA36 was discovered around same time as the c9 form of ALS/FTD in 2011. The ALS/FTD discovery has since formed the basis for myriad studies implicating these toxic protein aggregates as contributing drivers of disease in ALS and frontotemporal dementia (FTD).
"The genetic overlap was known, however the rate limiting step in studying SCA36 has been the availability of patient samples," says McEachin.
He traveled to Spain and Japan where there are known pedigrees of SCA36 patients to learn more about the disorder. To obtain SCA36 patient samples, the authors reached out to neurologists Maria-Jesus Sobrido, MD and Manuel Arias, MD at the Hospital Clínico Universitario in Santiago de Compostela, Spain, and neurologist Koji Abe, MD of Okayama University in Japan.
Emory University's ALS Clinic, directed by Jonathan Glass, MD, a close collaborator on this study, is the largest in the southeast and has curated a substantial biorepository of C9orf72 ALS and FTD biospecimens, including those used in this study.
"The study would not have been possible without the interest of these clinicians and willingness of their patients to participate," McEachin says. "We are truly grateful for their collaboration as this study wouldn't have been possible otherwise."
The authors also worked in collaboration with Leonard Petrucelli, Ph.D., and Tania Gendron, Ph.D. at Mayo Clinic Jacksonville, who developed specialized antibodies to analyze the nonsensical protein products. Wilfried Rossoll, Ph.D. now at Mayo Clinic, formerly at Emory, was also a collaborator.
In a separate paper released the same day in Cell Reports, these finding were further supported by similar observations in a novel mouse model of SCA36 generated by Leonard Petrucelli, Wilfried Rossoll and colleagues at Mayo. McEachin and Bassell were also co-authors on this manuscript.
A new technology for treating diseases with expanded DNA repeats—including Huntington's disease and myotonic dystrophy, as well as some forms of ALS and SCA—is emerging. "Antisense oligonucleotides" can be delivered to the nervous system and shut down production of the toxic proteins. The Emory ALS clinic is participating in some of the ASO clinical trials.
"We think our analysis will guide future research into potential treatments, for example, by developing drugs that can best target these related aggregation-prone molecules," Bassell says.
More information: Zachary T. McEachin et al, Chimeric Peptide Species Contribute to Divergent Dipeptide Repeat Pathology in c9ALS/FTD and SCA36, Neuron (2020). DOI: 10.1016/j.neuron.2020.04.011