Gene yields insights into the causes of neurodegeneration

Across the globe, approximately 50 million people are living with dementia. The two most common forms are Alzheimer's disease and frontotemporal lobar degeneration (FTLD), which develop when neurons in specific parts of the brain stop functioning—triggering memory loss and other behavioral or personality changes.
Without a cure, the World Health Organization predicts that number could rise by as many as 10 million cases per year. However, predicting the onset of these diseases is tricky because neurodegeneration can start years before people present any outward symptoms.
Cornell researchers including Fenghua Hu, associate professor in the Department of Molecular Biology and Genetics and member of the Weill Institute for Cell and Molecular Biology, are taking a closer look at the factors that cause Alzheimer's, FTLD and similar diseases. Hu's latest study, "A role of the frontotemporal lobar degeneration risk factor TMEM106B in myelination," was published June 23 in the journal Brain.
"I want to have a better understanding of the molecular and cellular mechanisms of neurodegeneration," Hu said. "I hope that our research can facilitate therapeutic development of treatment options for patients suffering from neurodegenerative diseases and other brain disorders."
Her team started by investigating a specific gene, called TMEM106B, which had been previously identified as a risk factor for several neurodegenerative diseases, including Alzheimer's and FTLD. Researchers also knew that a particular mutation in that gene caused a neurological defect known as hypomyelinating leukodystrophy, which creates a myelin deficit in the brain, leading to the deterioration of both motor skills and mental acuity.
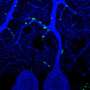
Myelin is the fatty tissue that wraps around nerve fibers, or axons, in the nervous system. Like insulation, this tissue forms a sheath that surrounds the nerve fibers, protecting them from damage and allowing electrical impulses to be quickly transmitted along the nerve.
Hu wanted to see exactly how that one mutation in TMEM106B could cause so much damage. She also wanted to learn more about how the gene regulates the formation and maintenance of the myelin sheath under normal conditions.
"We found that the mutation associated with the disease is a loss-of-function mutation," Hu said.
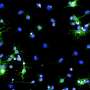
This distinction is critical since the Hu lab saw that TMEM106B is expressed in the cells that are responsible for forming the protective myelin tissue. Those cells are called oligodendrocytes, and within them, TMEM106B resides in the lysosome—a tiny organelle that acts as a cellular recycling center.
Like the stomach, the lysosome must maintain a specific pH to keep its enzymes active. As oligodendrocytes build the myelin sheath, lysosomes remove any extraneous materials. They can also store myelin's main membrane protein and deposit it in areas surrounding the nerve fibers.
Hu's team discovered that the TMEM106B mutation prevented the gene from regulating both the pH inside the lysosome and the movement of the lysosome itself—inhibiting the oligodendrocytes' ability to build compact myelin layers.
Using a mouse model, Hu also noticed that an overall TMEM106B deficiency led to abnormal lysosome movement within the oligodendrocytes. This created defects in the myelin sheath, and the team observed behavioral changes, including poor motor coordination.
Additional research will examine the exact mechanism by which TMEM106B regulates lysosome function and will demonstrate how the mutation leads to the known neurological defects.
"We want to explore whether the gene's regulation of myelination contributes to its association with neurodegenerative diseases, including Alzheimer's, FTLD and other age-related dementia," Hu said.
More information: Tuancheng Feng et al. A role of the frontotemporal lobar degeneration risk factor TMEM106B in myelination, Brain (2020). DOI: 10.1093/brain/awaa154