Blocking enzyme's self-destruction process may mitigate age-related diseases

Stopping the cannibalistic behavior of a well-studied enzyme could be the key to new drugs to fight age-related diseases, according to a new study published online in Nature Cell Biology. For the first time, researchers in the Perelman School of Medicine at the University of Pennsylvania show how the self-eating cellular process known as autophagy is causing the SIRT1 enzyme, long known to play a role in longevity, to degrade over time in cells and tissue in mice. Identifying an enzymatic target is an important step that may lead to new or modified existing therapeutics.
"Blocking this pathway could be another potential approach to restore the level of SIRT1 in patients to help treat or prevent age-related organ and immune system decline," said first author Lu Wang, Ph.D., a postdoctoral researcher in the lab of Shelly Berger, Ph.D., a professor of Cell and Developmental Biology in the Perelman School of Medicine and a professor of Biology in the School of Arts and Sciences at Penn. Berger also serves as senior author on the paper.
"The findings may be of most interest to the immune aging field, as autophagy's role in SIRT1 in immune cells is a concept that hasn't been shown before," Wang added. "Exploiting this mechanism presents us with a new possibility of restoring immune function."
Cells are like leaky faucets, dripping away levels of proteins and enzymes, such as SIRT1, as the body ages, which can lead to chronic diseases, organ decline, and weaker immune responses to infections. New ways to stop these leaks and replenish SIRT1 have been demonstrated, including by cardiovascular researchers at Penn Medicine, but this is the first study to show autophagy's role in that degradation during senescence—a natural process in which cells stop creating new cells—and aging.
SIRT1 is crucial for cell metabolism and immune responses, researchers have known, and has been shown to extend lifespan when overexpressed.
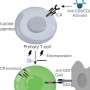
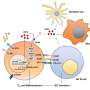
To determine the mechanism of SIRT1 loss during senescence, the researchers first ruled out it was driven by mRNA synthesis and stability, important factors in the control of gene expression, using RNA sequencing techniques on mouse cells. Instead, through further experiments, they found that "knocking out" the autophagic protein Atg7 in senescent cells left SIRT1 levels in place, indicating the autophagic pathway, and not proteasomes—the other recycling factory of the body—played a role in the loss of the enzyme. Immunofluorescence staining also showed that another autophagy protein, LC3, drives the loss of SIRT1 in senescent cells and tissue.
Treating mice with various drugs further supported autophagy's role. A proteasome inhibitor—which blocks the breakdown of proteins in the cell—failed to restore SIRT1 protein in senescent cells and tissue, while treatment with Lys05, an autophagy inhibitor, rescued the loss of SIRTI, supporting that the enzyme is degraded through lysosomes. Lysosomes are the "stomach" of cells that help break down larger waste materials.
To determine autophagy's role in SIRT1 in immune cells, the researchers treated human donor CD8 T cells with low-dose Lys05 and a proteasome inhibitor, and found that only Lys05 increased SIRT1 levels. The results, the authors said, indicate that SIRT1 is degraded at least in part through the autophagy-lysosome pathway during T cell aging in humans—a mechanism that could inform the reprograming of aged immune cells.
Next, the researchers will further explore the LC3 and SIRT1 interaction in preclinical studies and better characterize the signaling pathway to block it.
"Stabilizing SIRT1 protein level by interrupting this interaction could be a new direction for the design of anti-aging compounds," the authors said.
More information: Caiyue Xu et al. SIRT1 is downregulated by autophagy in senescence and ageing, Nature Cell Biology (2020). DOI: 10.1038/s41556-020-00579-5