Found: A genetic link to molecular events that precede symptoms in Alzheimer's disease
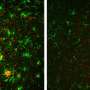
 and GGA3 (green) in a wild-type neuron. Credit: S. Lomoio et al., Science Translational Medicine (2020)")
Researchers at Tufts University School of Medicine have discovered a molecular mechanism that causes a "traffic jam" of enzymes traveling up and down neuronal axons, leading to the accumulation of amyloid beta—a key feature and cause of Alzheimer's disease. The enzyme, BACE1, gets backed up, causing the axons to clog and swell because of the increased production of the toxic amyloid protein.
The study, published today in Science Translational Medicine, reports that a human mutation more prevalent in African American patients with late onset Alzheimer's triggers a traffic jam of BACE1 in axons. Identifying this mutation is a key step in understanding the underlying molecular mechanisms of the disease and provides a possible strategy for early diagnosis and targeted treatments.
"In individuals with Alzheimer's disease, the onset of symptoms happens about 20 years after the first changes start to develop in the brain, making therapeutic intervention extremely difficult," said Giuseppina Tesco, professor of neuroscience at Tufts University School of Medicine and senior and corresponding author on the study. "So, we wanted to identify the mechanisms leading to the swelling of axons during the pre-symptomatic phase of Alzheimer's disease, which could in turn provide a way to detect the disease early and possibly treat it more effectively."
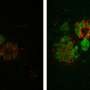
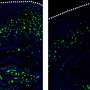
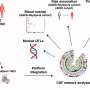
Tufts researchers previously identified a gene, Gga3, which helps regulate the traffic of BACE1, or beta-site APP-cleaving enzyme 1, along the axon. In the new study, the researchers found that when the Gga3 gene is mutated or missing in mice, their brains present the same distinctive traffic jam of BACE1 in swelling axons that are found in the postmortem brains of early stage patients with Alzheimer's disease. The researchers found that by disrupting the Gga3 gene, the traffic of BACE1 and other proteins along the axon is slowed or shut down. They also noted that a mutated or missing Gga3 leads to a severe accumulation of BACE1 in the axon, which results in axonal swellings both in cultured neurons and in a mouse model of Alzheimer's disease prior to amyloid deposition.
In multiple clinical trials, BACE inhibitors administered to patients with advanced disease who already had significant accumulation of amyloid beta protein and neuronal damage have been unsuccessful. The researchers asked whether application of the inhibitors at the earliest stages of disease might be more effective. They found that the inhibitors prevented swelling of axons in mice and even improved the two-way flow of BACE1. Their results suggest that earlier application of BACE1 inhibitors could be more effective at slowing the accumulation of amyloid beta protein.

Using datasets from the National Institutes of Health's National Institute of Mental Health and the Alzheimer's Disease Neuroimaging Initiative, the researchers discovered that mutations in Gga3 were more common among African Americans diagnosed with Alzheimer's disease than other populations. Although the sample size was small, the researchers believe this finding may provide a case for identifying early stage interventions and treatments for this group of patients.
"Our study provides a possible molecular explanation for the prevalence of axonopathy during the early stages of Alzheimer's disease, before the formation of amyloid plaques," said Tesco. "The mutation allowed us to determine that axonal alterations can be caused by accumulation of BACE1. Now an area of focus could be inhibiting BACE1 to prevent early axonal damage and perhaps this could also slow the development of amyloid plaques leading to disease."
The researchers note that the presence of neurofilament light chain (NfL) in blood plasma is a marker for axonal damage, and could be used to identify the best timing for use of BACE inhibitors to prevent or slow the progression of Alzheimer's disease during its early pre-symptomatic stages.
More information: S. Lomoio el al., "Gga3 deletion and a GGA3 rare variant associated with late onset Alzheimer's disease trigger BACE1 accumulation in axonal swellings," Science Translational Medicine (2020). stm.sciencemag.org/lookup/doi/ … scitranslmed.aba1871