Researchers reveal mechanism for α-syn deposition in familial Parkinson's disease

Amyloid deposition of α-synuclein (α-syn) is a hallmark of Parkinson's disease (PD). α-Syn fibrillation and cell-to-cell transmission in the brain play an essential role in disease progression. To date, eight single-point mutations of SNCA have been identified in familial PD (fPD), which feature early-onset, severe and highly heterogeneous clinical symptoms.
Previously, Prof. Liu Cong's group from the Shanghai Institute of Organic Chemistry (SIOC) of the Chinese Academy of Sciences revealed that α-syn mutations including E46K, A53T form different fibril structures that are distinct from wild-type (WT) α-syn fibrils. Whether and how hereditary mutations-induced fibril polymorphism contributes to the early-onset and exacerbated pathology in fPD remains to be elucidated. More importantly, most fPD patients are heterozygous for SNCA mutations, which leads to another critical question: could mutant fibrils cross-seed WT α-syn to orchestrate neuropathology in fPD patients?
In a recent study published in PNAS on May 11, Prof. Liu Cong and Prof. He Kaiwen's groups from SIOC revealed the structural basis underlying the cross-seeding between the WT and fPD mutant α-syn, underscoring the importance of fibril structure in determining α-syn neuropathology, and providing mechanistic understanding for the pathology of E46K-associated fPD.
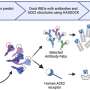
In this study, the researchers found that human E46K (hE46K) and human WT (hWT) fibril strains induced α-syn aggregates with distinct morphologies in mice brain. Mice injected with the hE46K strain induced more endogenous α-syn aggregation and early-onset motor deficits compared to the mice injected with the hWT strain.
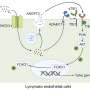
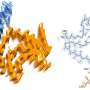
Notably, the hE46K fibril cross-seeded hWT α-syn, and induce hWT to formed hWTcs fibril stain which replicated the structure and seeding capability of the hE46K template both in vitro and in vivo.
These results suggest that the E46K fibril could propagate its structure as well as the seeding properties to the WT monomer so as to amplify the α-syn pathology in fPD (Fig. 1).
This work suggests that the structural and pathological features of mutant strains could be propagated by the WT α-syn in such a way that the mutant pathology would be amplified in fPD.
More information: Houfang Long et al. Wild-type α-synuclein inherits the structure and exacerbated neuropathology of E46K mutant fibril strain by cross-seeding, Proceedings of the National Academy of Sciences (2021). DOI: 10.1073/pnas.2012435118