A leap in understanding of hypertrophic cardiomyopathy

Hypertrophic cardiomyopathy (HCM) is the most common of all genetic heart diseases and is the leading cause of sudden cardiac death. It is characterized by an abnormal thickening of the heart muscle, which over time can lead to cardiac dysfunction, and ultimately, heart failure.
A paper published in the Proceedings of the National Academy of Sciences (PNAS) and co-authored by Beth Pruitt, a UC Santa Barbara professor of mechanical engineering and the director of the campus's Institute for BioEngineering, describes the results of a complex long-term collaboration that included researchers at Stanford University, the University of Washington, and the University of Kentucky. The study has led to new understanding of how genetic mutations play out at the cellular level to cause HCM, and new perspectives on how to prevent it.
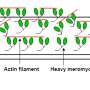
In their paper, the authors explain that more than one thousand genetic mutations that cause HCM have been identified. The majority of them are found in genes that encode sarcomeric proteins, the structural building blocks of heart muscle responsible for generating and regulating contraction and relaxation. Roughly a third of the mutations are located in beta cardiac myosin, the primary protein that drives contraction of the heart cells. Contraction of the heart muscle, and every other muscle in our bodies, results from a process in which the motor protein myosin "walks" along a chain of actin molecules, a process known as the cross-bridge cycle. During this process, chemical energy in the form of ATP is converted to mechanical energy, ultimately leading to cardiac contraction.
Prior to a contraction, the head of one strand of an intertwined two-strand myosin molecule is tucked up against an actin molecule. Muscle contraction is initiated when a molecule of ATP, known as the "energy currency" of biological systems, binds to the myosin head. The myosin head, and the attached ATP, then detaches from the actin, initiating hydrolysis of the ATP, which is transformed into ADP plus a phosphate group. That process releases energy that "cocks" the myosin protein into a high-energy state and changes the shape of the myosin so that it is ready to crawl along the actin. At that point, the phosphate is released from the myosin, causing the myosin to push on the actin and release the phosphate, which leads the myosin to walk to the next chain of actin and contract the muscle. All of this, involving millions of heads of myosin walking across actin in steps that take microseconds to complete, must occur at the proper rate in order to maintain heart health.
Because HCM is often observed in patients having mutations in the beta cardiac myosin protein, it had been hypothesized that HCM mutations cause a cascade of events that manifest, ultimately, in damage to the heart itself. This study put that idea to the test, focusing on a single mutation, P710R, which dramatically decreased in vitro motility velocity—the rate at which the myosin motor walks on actin—in contrast to other MYH7 mutations, which led to increased motility velocity.
The overarching research question of this project was to learn how a mutation linked to heart disease in patients changes heart function at a cellular level.
The team used CRISPR technology to edit human induced pluripotent derived stem cell cardiomyocytes (cells responsible for heart contraction) by inserting the P710R mutation into them. Pruitt leads the stem cell bank at UCSB, where "clean" cell lines, having no genetic abnormalities, are maintained and reproduced for university researchers. Such clean, mutation-free lines provide a perfect benchmark for comparison with cells to see very precisely the effects of the P710R mutation. For example, the research team is now testing the effects of different mutations linked to heart disease in the same genetic background.
"You can have ten people with the same gene mutation in this protein, and they can have varying degrees of clinical significance, because the rest of their genome is different; that's what makes us individuals," Pruitt said. "These lines let us examine what is a result of the genetic mutation. By comparing the effect of different mutations, we can begin to tease apart how these changes lead to HCM. It allows us to look closely at how and why the cells adapt to the mutation in that way, and to get data and relate it to the thickness of the heart wall and all the other things that happen downstream."
This research began nearly 15 years ago, while Pruitt was still at Stanford, and led to this collaborative paper. Now CRISPR technology enables researchers to design cells expressing specific mutations that are linked to cardiac diseases, and then assess molecular and functional changes to determine the cellular impact of individual mutations that have been identified in patients with HCM. These studies will provide a mechanistic understanding of how individual mutations at the molecular level translate to HCM in patients.
In this project, once the mutation was introduced, the cells were assayed in a collaboration between the Pruitt lab (UCSB) and the Bernstein lab (Stanford University), using traction force microscopy, an assay that allows simultaneous observation of a beating cell and the force it generates. The Spudich lab (Stanford), led separate studies of the same mutated protein at the molecular level using an optical trap, in which light pressure is applied to control precisely the location and force of an actin "dumbbell" held between beads as myosin heads walk along the actin, to measure myosin's power cycle. The assay revealed that the P710R mutation reduced the step size of the myosin motor (i.e. the length of each step) and the rate at which the myosin detaches from actin.
In a collaboration with University of Kentucky researcher Kenneth Campbell, these observations were then compared to a computational model of how the myosin motors interact in the cell to generate force. The results confirmed a key role for regulation of what is called myosin's "super-relaxed state." As Pruitt explained, "Myosin heads spend a lot of time in a super-relaxed state, referring to when it is unbound from actin. Any mutation or drug that shifts how long or how strongly myosin motors are bound to actin will change the cell force production and change downstream signaling events that drive remodeling and growth or hypertrophy."
The P710R mutation in this study was found to destabilize the super-relaxed state. As a result, more myosin heads are bound to actin in cells that harbor the mutation, which explains the increase in force that was observed in those cells.
For Pruitt, a key takeaway from the work, beyond the important scientific findings, is the value of sustained collaboration. "The scales that the paper covers are not typically the subject of research in any one lab or even any two labs," she said. "That's why the paper has so many authors, including several students and postdocs working with me, James Spudich and Daniel Bernstein.
"It's significant scientifically but also satisfying in that this level of integration makes it possible to test this idea across multiple scales. It's been fun to work across these labs and these skills on such an extensive, multidisciplinary collaboration, and to see that the power of molecular measurements and computation, and the cell-derived measurements that allow us to genetically engineer and dissect out a single mutation," said Pruitt. "This is really phenomenal, to test directly how a particular mutation introduces changes that lead to HCM."
As a result of this collaboration, Pruitt added, "We can understand what goes on at the cell level. Then we can start to develop models and identify next-generation drug therapies. Instead of just identifying the symptoms, we can look at the mechanisms that underlie the dysfunctions and then address those at the cell level before it turns into a disease."
More information: Alison Schroer Vander Roest et al, Hypertrophic cardiomyopathy β-cardiac myosin mutation (P710R) leads to hypercontractility by disrupting super relaxed state, Proceedings of the National Academy of Sciences (2021). DOI: 10.1073/pnas.2025030118