Researchers identify the biological mechanism causing nerve destruction in the motor neuron disease ALS

A research group from the Sackler Faculty of Medicine and the Sagol School of Neuroscience at Tel Aviv University uncovered, for the first time, the biological mechanism causing nerve destruction in the neurodegenerative disease ALS. The groundbreaking study, led by Prof. Eran Perlson and the doctoral students Topaz Altman and Ariel Ionescu, suggests that the course of this fatal disease can be delayed and even reversed in its early stages. It was conducted in collaboration with Dr. Amir Dori, director of the clinic for neuro-muscular diseases at Sheba Medical Center. The results of the study were published in Nature Communications.
ALS is the most common type of motor neuron disease, causing paralysis and muscle atrophy. One out of every 400 people will have the disease, and yet it has no effective cure. ALS patients gradually lose their ability to control their voluntary muscle movements, leading to complete paralysis and eventually lose the ability to breathe independently. The average life expectancy of ALS patients is currently only about three years.
"To this day it is unclear what causes the disease", explains Prof. Perlson. "Only about 10% of the patients carry a familial background with known genetic mutations, but the remaining 90% are a mystery. The paralysis caused by the disease results from damage to the motor neurons, which leads to the degeneration nerve endings and to the loss of muscle innervation. This consequently leads to the degeneration of the nerve and the death of motor neurons in the spinal cord, however until now we could not understand the basic biological mechanism causing the initial damage behind this vicious cascade".
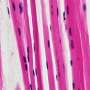
 whereas the left picture was taken at high magnification (600x). Credit: Tel Aviv University")
To solve the mystery, the Tel Aviv University researchers focused on a protein called TDP-43, which had been shown in earlier studies to accumulate in unusual amounts and localization in the brains of about 95% of all ALS patients. Prof. Perlson and his team revealed a novel biological link between the protein's accumulation and the degeneration of the synapses between the motor neuron endings and the muscles, called neuromuscular junctions, which translate neural commands into physical movements. In muscle biopsies taken from ALS patients the researchers found that the toxic protein accumulates also in high proximity to these neuromuscular junctions during the early stages of the disease and before patients develop any serious symptoms. In a series of experiments performed by the researchers, both in cells of ALS patients and in genetically modified model animals, they discovered that the accumulation of the TDP-43 protein in the neuromuscular junction inhibits the ability to locally synthesize proteins that are essential to mitochondrial activity, which provides the power of fundamental cellular processes. The dysfunction of mitochondria in nerve terminals leads to neuromuscular junction disruption and ultimately to the death of the motor neurons. "It's important first to understand the spatial complexity of motor neurons", says Prof. Perlson.
"The motor neurons are found in the spinal cord and need to reach every muscle in the body in order to operate it. One can imagine, for example, an extension cable coming out of the spinal cord and reaching the muscles in the little toe in our foot. These extensions can be as long as one meter in adults and are called axons. In earlier studies, we have shown that to maintain this complex organization motor neuron axons require an increased amount of energy, particularly in the most remote parts, the neuromuscular junctions. In our current study, we focused on a pathological change in TDP-43 protein that takes place in these axons and at neuromuscular junctions. In a normal motor neuron, this protein is mainly found in the nucleus. We showed that in ALS this protein exits the nucleus and accumulates throughout the entire cell and particularly in the neuromuscular junction. As the function of motor neurons depends on these neuromuscular junctions located on the remote end of the "extension cable", we realized that this finding could be of critical importance. We discovered that the accumulations formed by the TDP-43 protein in neuromuscular junctions trap RNA molecules and prevent the synthesis of essential proteins to mitochondrial function. Mitochondria are organelles found in cells and are the main energy providers for numerous cellular processes, including neural transmission. The condensation of TDP-43 protein in neuromuscular junctions results in a severe energy depletion, prevents mitochondrial repair, and consequently leads to the disruption of these junctions, degeneration of the entire 'extension cable' and to the death of motor neurons in the spinal cord."
In order to confirm their findings, the Tel Aviv University researchers decided to use an experimental molecule recently published by a group of researchers from the US for developed for another purpose—enhancing neural regeneration after injury by the disassembly of protein condensates in neural extensions. The researchers proved that this molecule could also disassemble the axonal TDP-43 protein condensates in cells from ALS patients, and that this process improved the ability to produce essential proteins, enhanced mitochondrial activity, and prevented neuromuscular junction degeneration. Additionally, in the model animals, the researchers showed that by reversing TDP-43 accumulation in nerves and neuromuscular junction enabled recovery of degenerated neuromuscular junctions—and to rehabilitate the diseased model animals almost completely.
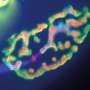
 by the motor nerve (in red). Center: degenerated neuromuscular junction in diseased model mice, showing the disruption of the motor nerve (red) and the reduction in the receptor area on the muscle in response to the disruption (green). Left: neuromuscular junction in diseased model mice after clearing the TDP-43 protein from nerve extensions. Image demonstrated the renewed innervation the receptors on the muscle (green) by the motor nerve extension (red). Credit: Tel Aviv University")
"The moment we induced the disassembly of TDP-43 protein condensates, the nerves' ability to produce proteins was recovered, particularly the synthesis of proteins essential to mitochondrial activity. All this made it possible for the nerves to regenerate", summarizes Prof. Perlson. "We were able to prove, through pharmacological as well as genetic means, that motor nerves can regenerate—and that patients can have hope. In fact, we located the basic mechanism, as well as the proteins responsible for the disruption of the nerves from the muscles and for their degeneration. This discovery can lead to the development of new therapies that could either dissolve the TDP-43 protein condensates or increase the production of proteins essential to mitochondrial function, and thereby heal the nerve cells before the irreversible damage that occurs in the spinal cord. We are tackling the problem on the other end—in the neuromuscular junction. And if in the future we could diagnose and intervene early enough, maybe it will be possible to inhibit the destructive degeneration in ALS patients' muscles".
The study is an international collaboration with leading scientists from Germany, France, England and the US, with the assistance of Tal Gardus Perry and Amjad Ibraheem from Prof. Perlson's laboratory.
More information: Topaz Altman et al, Axonal TDP-43 condensates drive neuromuscular junction disruption through inhibition of local synthesis of nuclear encoded mitochondrial proteins, Nature Communications (2021). DOI: 10.1038/s41467-021-27221-8