Neurodegeneration can be studied long before symptoms arise

Many changes take place in the brain long before symptoms appear. This has been shown in mice in two studies of prion diseases in which the brain gradually deteriorates. The results suggest that changes can be studied extremely early in the disease process, which is important if we are to develop treatments. The studies, published in PLOS Pathogens and Life Science Alliance, have been carried out by researchers at Linköping University, Sweden.
Prion diseases are rare, incurable diseases that cause the brain to deteriorate. Two well-known examples are mad-cow disease in cattle and scrapie in sheep. In the 1990s, people who ate meat from cattle infected with mad-cow disease were affected by a variant of the disease.
"Prions are remarkable entities. They are infectious agents that consist solely of a specific protein. The normal form of the prion protein is found in all mammals, but it can undergo a strange transformation and become toxic. This transformation sets off a cascade of changes that converts normal protein molecules into the toxic variant," says Lech Kaczmarczyk, principal research engineer in the Department of Biomedical and Clinical Sciences (BKV) at Linköping University, and principal author of one of the studies.
Prion diseases can be spread to organisms by infectious prions. But they can also arise following genetic changes (mutations) in the gene that codes for the prion protein. The area of the brain affected depends on the location of the mutation in the gene. This can happen even though the normal prion protein is present in many cell types and many areas of the brain. This surprising property is known as "selective vulnerability," and it can be seen in other, more common, diseases that affect the nervous system (neurodegenerative diseases). In people with Parkinson's disease, for example, we see damage in the parts of the brain that control body movement, while in Alzheimer's disease degeneration of other parts of the brain cause dementia and cognitive problems.
Researchers wanted to identify the underlying causes of selective vulnerability that leads to different diseases giving different symptoms. One major challenge for researchers is that the parts of the brain involved have already suffered damage when the disease starts to become manifest. This makes it difficult to investigate how the process started.
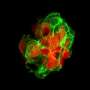
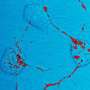
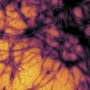
The researchers who conducted the current studies looked at disease models of various prion diseases in mice. The researchers used new methods that allow them to discover disease mechanisms long before symptoms appear. There are no visible changes in brain activity, no changes in animal behavior, and the brain tissue appears completely normal, but the researchers know that the mice are destined to develop the disease, and they can predict when it will happen.
"Many researchers and clinicians take the position that therapies for neurodegenerative diseases must be applied at very early stages, even before clinical signs emerge, to provide any benefit. This is why it's important to understand what is happening in the earliest stages of the disease," says Walker Jackson, associate professor at BKV, Linköping University, and leader of the studies.
In one of the studies, researchers have looked at how different cell types react when infected with prions, while in the other they have studied two genetic prion diseases: fatal familial insomnia and Creutzfeldt-Jakob disease (CJD). FFI is characterized by severe insomnia, while the principal effect of CJD is dementia and impaired cognitive functions. Even though these two diseases show different symptoms in their later stages, the researchers found similarities in the early stages, before symptoms appear.
"In our studies, we see coordinated changes in gene expression in the very early stages of the diseases, as if the cells are trying to change something in their biology to fight off the disease. Some researchers consider changes in gene expression patterns during disease to be a dysfunction, but our results suggest that these are coordinated cellular responses," says Walker Jackson.
In the first study, the expression patterns of so many genes were altered in the early stages of the diseases, just before symptoms emerge, that it would be impossible to select a treatment target. This discovery may help to explain why many drug studies did not yield the desired treatment results; they focused on a single gene or target and missed the other problems in the cell.
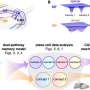
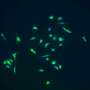
Cells are continuously producing new proteins, as determined by which genes are active at any one time. They are produced in organelles in the cell known as "ribosomes." For both studies the researchers used a method that allowed them to follow the ribosomes in one specific cell type, and thus obtain an idea of what is happening in the cell at that particular moment.
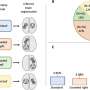
In their second study, they looked at six cell types of the two genetic prion diseases, caused by different mutations in the prion gene.
"We were surprised when it turned out that the same cell type reacts most strongly in the two diseases, and that the cells react in essentially the same way. So even though the two diseases are very different in their later stages, there may be underlying similarities that are interesting targets for treatment in the long term," says Susanne Bauer, doctoral student in BKV and principal author of the second study, published in Life Science Alliance.
The cell type in question is a nerve cell that expresses somatostatin, and it has not been previously studied in prion disease research in any detail. Earlier studies have, however, suggested that this cell type can be affected in the early stages of other neurodegenerative diseases.
The researchers also saw that the cellular changes in the early stages of disease were completely different in genetic prion diseases from those in infectious prion diseases. This shows that even if it is fundamentally the same prion protein that is defective, there are large molecular differences between prion diseases of different origins. The researchers are now continuing their studies and will use the same methods to look at other, more common, neurodegenerative diseases.
More information: Lech Kaczmarczyk et al, Distinct translatome changes in specific neural populations precede electroencephalographic changes in prion-infected mice, PLOS Pathogens (2022). DOI: 10.1371/journal.ppat.1010747
Susanne Bauer et al, Translatome profiling in fatal familial insomnia implicates TOR signaling in somatostatin neurons, Life Science Alliance (2022). DOI: 10.26508/lsa.202201530