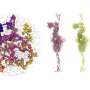Adapting language models to track virus variants
 The embeddings from GenSLMs are visualized with t-distributed stochastic neighbor embedding (t-SNE) and each gene sequence is represented as a dot in the 2D plot. We paint each sequence by its variant ID—although we have more than 515 PANGO (Rambaut et al., 2020) lineages represented in the data, we only show those with WHO designated labels. (B) The latent space can also be painted with the MAFFT-determined alignment score (Yamada et al., 2016) with respect to an omicron genome; clustering in the distance measures is clearly visible. Visualizing the sequence log-likelihood (blue bar) and the cross-protein attention (orange lines) from (C) Delta and (D) Omicron SARS-CoV-2 strains highlights how different the coevolutionary patterns are in these lineages. It is interesting to note that while the Spike protein from delta strain shows coupling to nsp3, nsp5, and other proteins, these couplings are not observed in the omicron strain. Credit: bioRxiv (2022). DOI: 10.1101/2022.10.10.511571")
Scientists from the U.S. Department of Energy's (DOE) Argonne National Laboratory and a team of collaborators have won the 2022 Gordon Bell Special Prize for High Performance Computing-Based COVID-19 Research for their new method of quickly identifying how a virus evolves. Their work in training large language models (LLMs) to discover variants of SARS-CoV-2 has implications to biology beyond COVID-19.
A form of artificial intelligence (AI), LLMs are generally meant to summarize and translate texts or predict what words might come next based on what the model learned in an initial training stage. For instance, an LLM can be trained—with the help of gigantic language datasets—to translate text from English to Spanish.
The researchers who won this year's award leveraged Argonne's powerful supercomputing and AI resources to develop and apply LLMs toward tracking how a virus can mutate into more dangerous or more transmissible variants.
When a virus evolves, it mutates into new variants that can be similar to past variants or even more deadly than previous iterations. When a particular variant is considered more dangerous or harmful, it is labeled as a variant of concern (VOC). Discovering these VOCs quickly and efficiently can save lives by providing scientists with time to design and develop effective vaccines and treatment strategies.
Existing methods to track these variants can be slow. To solve this problem, computational biologist Arvind Ramanathan and his colleagues at Argonne together with collaborators from the University of Chicago, NVIDIA, Cerebras Inc., University of Illinois at Chicago, Northern Illinois University, California Institute of Technology, New York University and Technical University of Munich set out to create a means of identifying VOCs. Their paper, "GenSLMs: Genome-scale language models reveal SARS-CoV-2 evolutionary dynamics," is the culmination of the team's findings.
"When the pandemic began, we had several of these really harmful variants of the virus, like the Delta variant," Ramanathan said. "It resulted in a large death toll. But Delta evolved as a consequence of certain mutations that were happening when the virus was facing the human hosts. It's a process of evolution of the virus inside of the human cell."
Their work resulted in the first genome-scale language model (GenSLM), which is a model that can analyze genes and rapidly identify VOCs. The model discussed in the paper was trained on data from the COVID-19 pandemic, and the hope is that models like this could potentially give health officials the tools they need to quickly respond to rising variants. GenSLM is the first whole genome-scale foundation model that can be altered and applied to other prediction tasks similar to VOC identification.
While these evolutionary variants may seem to crop up randomly to the human eye, tracking them is of the utmost concern. As such, the work of Ramanathan and his colleagues could seriously alter how we stay on top of viral outbreaks.
The language of evolution
Previous work demonstrated that LLMs based on the amino acid language of proteins can be used both to track the evolution of proteins and to design entirely new proteins with novel structure and function. However, Ramanathan points out that, to his knowledge, the research he and his colleagues performed was the first attempt at running an LLM-based model at the gene level.
"Large language models are key for achieving the AI for science vision across diverse science domains," said Venkatram Vishwanath, co-author of the study and data science lead at the Argonne Leadership Computing Facility (ALCF), a DOE Office of Science user facility.
That said, proteins are still two steps away from the core biological process that Ramanathan and his team were interested in. In the cell, genes are first transcribed onto something called messenger RNA (mRNA). This mRNA exits the cell's nucleus and makes its way into ribosomes, where the ribosomes synthesize proteins. In a sense, you could consider genes to be a message containing instructions on how to build proteins. And since proteins function somewhat similar to conversational messages, it would make sense to apply language models to define them.
While previous experiments had proven that language models were adept at explaining evolutionary changes at the protein level, scientists needed to go deeper if they wanted to identify VOCs at the gene level.
Machine learning played a pivotal role in this research, and the models needed information to learn about VOCs. The Bacterial and Viral Bioinformatics Resource Center web resources as well as the Houston Hospital System provided integrated data and analysis tools to support this work. The researchers analyzed 1.5 million high-quality SARS-CoV-2 complete genome sequences from the resource center and 16,545 total sequences from Houston to better understand the virus.
Previously, without this GenSLMs, VOCs needed to be identified by individually going through every protein and mapping each mutation to see if any mutations were of interest. This is incredibly labor- and time-intensive, and GenSLMs should help make this process easier.
The team has proven that these models can help advance biology research, and they now want to understand how far they can push the approach. Ramanathan believes that their work could lay the groundwork for a future pandemic observatory. He also suggests that protein engineering applications could come from this work, or even the modeling of entire organisms.
The right tools for the job
Powerful supercomputing assets were vital in the success of this work. The researchers used both Polaris, a Hewlett Packard Enterprise system, and the Cerebras CS-2 AI platform at the ALCF. This research also relied on NVIDIA's Selene supercomputer. While Polaris and Selene are powerful supercomputers accelerated by GPUs (graphics processing units), the CS-2 system is different. The CS-2 AI-accelerator system, part of the ALCF AI Testbed, is highly optimized for learning-based tasks.
"Polaris is the new ALCF supercomputer with four GPUs on a single node, and we have 560 of these nodes," said Ramanathan. "This really helps us scale the end-to-end workflow, including the training process, across multiple nodes in a much more convenient manner. And because of the amount of memory and node-local storage that is available on a single node, we can load or we can basically stage the data in certain ways that let us utilize the entire machine's power for doing these types of complex calculations."
Given the critical time crunch to obtain results, the team also relied on Cerebras CS-2 machines to aid in their work in addition to the Polaris and Selene systems. Specifically, they utilized the Cerebras Wafer-Scale Engine and ended up requiring both a single CS-2 machine as well as a cluster of 16 CS-2 to achieve the desired accuracy and perplexity results in less than a day.
"A key challenge in this problem is dealing with long sequence lengths and tackling these foundation models at the scale of the viral genome," said Ramanathan. "This process can benefit from systems with large memory capabilities, such as the CS-2 system architecture with their Memory-X and Swarm-X infrastructure, and this makes it easier to load and train on these very long sequences."
"Resources such as Polaris, CS-2 systems and the various AI accelerator systems at the ALCF AI Testbed are helping us advance the use of these models for scientific research," said Vishwanath.
More information: Maxim Zvyagin et al, GenSLMs: Genome-scale language models reveal SARS-CoV-2 evolutionary dynamics, bioRxiv (2022). DOI: 10.1101/2022.10.10.511571