Scientists define independent subtype of idiopathic multicentric Castleman disease
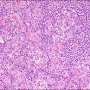
 Interfollicular areas are expanded and germinal centers (GCs) appear hyperplastic (arrowheads) (H&E, 10×). (B) The sheet-like proliferation of mature plasma cells in the interfollicular areas and hemosiderin deposition are observed (arrow heads) (H&E,40×). (C) Numerous plasma cells are observed in the interfollicular areas (CD138, 20×). (D) Hemosiderin deposition is observed (arrowheads) (Berlin blue staining, 40×). This case was scored: vascularity grade 0, plasmacytosis grade 3, regressed GC grade 2, and hyperplastic GC grade 2. Credit: International Journal of Molecular Sciences (2022). DOI: 10.3390/ijms231810301")
Multicentric Castleman disease (MCD) comprises a heterogenous group of rare disorders that exhibit generalized symptoms such as swelling of lymph nodes, anemia, fever, and fatigue. The causative factors underlying MCD include Kaposi sarcoma-associated herpesvirus (KSHV)/human herpesvirus 8 (HHV8) infections.
KSHV/HHV8-negative MCD without association with the POEMS (polyneuropathy, organomegaly, endocrinopathy, M proteins, and skin changes) syndrome are termed "idiopathic," i.e., diseases with no identifiable cause, and are thus referred to as "iMCD". At present, specific clinical manifestations or diagnostic criteria have not been identified for iMCD, which often results in treatment delay.
iMCD is further classified as iMCD-TAFRO (thrombocytopenia, anasarca, fever, reticulin fibrosis, and organomegaly) and iMCD-NOS (not otherwise specified). With additional research efforts, iMCD-NOS, which is highly heterogenous, could be classified into various subtypes.
Interestingly, a disease known as idiopathic plasmacytic lymphadenopathy (IPL), which is characterized by hypergammaglobulinemia, i.e., high levels of serum immunoglobulins, and histologically mature plasma cell proliferation in the lymph nodes, is considered a part of iMCD-NOS due to their clinicopathological similarity. However, till date, no study has validated if IPL has distinct clinicopathologic features compared to other iMCD-NOS.
This motivated a team of researchers led by Assistant Professor Asami Nishikori, including Associate Professor Midori Filiz Nishimura, and Professor Yasuharu Sato from Department of Molecular Hematopathology, Okayama University Graduate School of Health Sciences, Japan to conduct a thorough clinicopathological examination of IPL and others in iMCD. Their findings were published in the International Journal of Molecular Sciences.
"We wanted to determine whether IPL has distinct and uniform clinicopathologic features compared to other subtypes (non-IPL) in iMCD-NOS," says Assist. Prof. Nishikori while discussing the motivation behind the study.
The team analyzed lymph node specimens derived from 42 Japanese patients with lymph node involvement of iMCD-NOS, who met the consensus diagnostic criteria of iMCD and were negative for KSHV/HHV8 infections. Upon further examination, the team classified 34 of the 42 patients as the IPL group and the remaining 8 as the non-IPL group.
On grading histological features such as vascularity, plasmacytosis (high proportion of plasma cells), atrophic and hyperplastic germinal centers (GCs), the team identified significant pathological differences between the specimens of the IPL and non-IPL groups. The IPL group demonstrated greater plasmacytosis and hyperplastic GCs compared to the non-IPL group. Conversely, the vascularity of the non-IPL group was higher than that of the IPL group.
Clinically, the IPL group exhibited higher platelet count and serum antibody (immunoglobulin G) levels, with lesser fluid retention in pleural and/or abdominal cavity. The frequency of disease-specific autoantibody detection was also different between the groups.
In addition, the clinical course and treatment responses in the two groups were also distinct. Upon treatment with tocilizumab, an anti-interleukin 6 receptor monoclonal antibody approved to treat iMCD in Japan, there was a significant improvement in the condition of patients with IPL. In contrast, patients with non-IPL responded poorly to the drug and showed progressed disease activity, requiring more intensive treatment.
These findings were consistent with those of previous studies that suggest that the clinical course of IPL disease progression is slower as compared to the non-IPL group, and with a superior response to anti-IL-6 agents. Discussing these findings, Assist. Prof. Nishikori states "Given the heterogeneity of non-IPL cases, we must try to identify its primary cause. Molecular analysis could lead to a better understanding of disease pathology and the development of subsequent targeted therapies."
To sum up, these findings have established IPL as a clinicopathologically uniform disease, which can be reclassified into an independent subtype of iMCD.
So, what are the future implications of these findings? "It is critical that patients get the most out of any treatment modules. Studies on iMCD subtype-specific diagnostic biomarkers, treatment, and follow-up are thus warranted in the future to improve the clinical outcomes for these patients," concludes Assist. Prof. Nishikori.
The paper is published in the International Journal of Molecular Sciences.
More information: Asami Nishikori et al, Idiopathic Plasmacytic Lymphadenopathy Forms an Independent Subtype of Idiopathic Multicentric Castleman Disease, International Journal of Molecular Sciences (2022). DOI: 10.3390/ijms231810301