This article has been reviewed according to Science X's editorial process and policies. Editors have highlighted the following attributes while ensuring the content's credibility:
fact-checked
peer-reviewed publication
trusted source
proofread
Using human stem cells to model a severe epilepsy syndrome and identify a potential targeted treatment

Mutations in the SCN3A gene cause a spectrum of neurological conditions collectively referred to as SCN3A-related neurodevelopmental disorders, which includes different types of epilepsy and brain malformations.
In a new study from researchers at Children's Hospital of Philadelphia (CHOP), human induced pluripotent stem cell (IPSC) technology and gene editing were used to model SCN3A disorders, establish mechanisms of abnormal neuronal function, and identify a pharmacologic agent that selectively blocks the channel that could represent a potential therapeutic option for these patients in the future.
The findings were recently published online by the journal Brain.
The SCN3A gene encodes the instructions to form a sodium channel, which is a protein complex that allows positively charged sodium ions into brain cells that activate neurons to carry electrical signals between brain cells throughout the brain and our nervous system.
However, missense variants—resulting from single base pair changes in DNA that lead to switched amino acids—can affect a sodium channel subunit called Nav1.3, leading to early onset and severe epilepsy as well as structural malformation in the development of the cerebral cortex of the brain.
Despite knowing which subunits and variants are associated with SCN3A-related neurodevelopmental disorders, the mechanisms of how they lead to disease symptoms were unclear.
To help unravel this mystery, researchers used human IPSC technology and CRISPR/Cas9 gene editing to compare human neurons harboring disease-associated SCN3A variants as well as so-called isogenic controls with normal sodium channels to study how SCN3A controls the electrical activity of early brain neurons and how SCN3A mutations alter the activity of developing neurons.
"SCN3A-related neurodevelopmental disorders are currently untreatable and are associated with high morbidity and mortality in pediatric patients, meaning that it is imperative to find effective therapies for this group of disorders," said Jerome Clatot, Ph.D., a research scientist in the Division of Neurology and co-first author of the study along with former postdoctoral fellow Quojie Qu and Penn Neuroscience Graduate Group student Julie Merchant.
"Understanding the mechanistic basis of how genetic variation in SCN3A leads to neurological disease could help us develop novel therapeutic approaches."
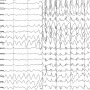
The researchers were able to develop forebrain-like neurons from IPSCs (iNeurons) that expressed the variant Nav1.3-mediated sodium ion currents as well as control iNeurons. With this method, the researchers found that the modified iNeurons expressing the variant had increased slowly-inactivating ("persistent") sodium ion currents that led to abnormal neuron firing patterns.
To test whether the Nav1.3 subunit might be a therapeutic target for potential treatments for this group of disorders, the researchers tested a Nav1.3-selective blocker. They found that after the blocker was applied, abnormal neuronal activity was restored to normal.
"This study is a nice use of human IPSC technology to model the effects of genetic variation in SCN3A while also revealing how Nav1.3 regulates the electrical activity of developing neurons," said senior study author Ethan Goldberg, MD, Ph.D., a pediatric neurologist and Director of the Epilepsy Neurogenetics Initiative (ENGIN) at CHOP. "Early results of the Nav1.3-selective blocker provide proof of principle supporting this general therapeutic approach towards SCN3A-related disorders, which we hope to explore in more detail in future studies."
More information: Guojie Qu et al, Targeted blockade of aberrant sodium current in a stem cell-derived neuron model of SCN3A encephalopathy, Brain (2023). DOI: 10.1093/brain/awad376