This article has been reviewed according to Science X's editorial process and policies. Editors have highlighted the following attributes while ensuring the content's credibility:
fact-checked
peer-reviewed publication
trusted source
proofread
New method reveals hidden genetic variations
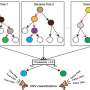
. As a consequence, reads that align onto these regions will have mapping qualities of 0 (when no masking is applied). To indicate this, reads are displayed white. Within Chameleolyser, reads are extracted and re-aligned onto a reference sequence in which R2 is masked. As a result, reads align uniquely onto R1 and will have mapping scores different from 0. This is indicated by representing them in gray. By applying a sensitive variant calling onto this masked alignment, Chameleolyser is able to identify single nucleotide variants and small indels (SNVs/Indels; green bullet in panel b). Nevertheless, the exact position of the variant remains ambiguous, hence we named them VAPs (variant with ambiguous position). In case R1 and R2 are identical in sequence, Chameleolyser limits the identification of homozygous deletions to events in which both R1 and R2 are deleted (panel c). Panels d, e and f illustrate the scenarios in which R1 and R2 are not completely identical (Seq. Id ≠ 100%). The three positions in which R1 differs from R2 are indicated with a colored bullet. Since reads that align onto these regions will have sufficiently good mapping qualities, the identification of regular SNVs/Indels does not pose a problem for standard data analysis pipelines. Nevertheless, SNVs/Indels that result from a gene conversion typically remain unidentified. By only considering the coverage profile of R1, an ectopic gene conversion and a deletion look identical (panels e and f). Chameleolyser also considers the coverage at locus R2. As a result, gene conversions can be distinguished from deletions. Indeed, in case of an ectopic gene conversion, reads that originate from the acceptor site will align onto the reference sequence of the donor site resulting in an increased sequencing coverage as opposed to the scenario where no gene conversion is present. Credit: Nature Communications (2023). DOI: 10.1038/s41467-023-42531-9")
Many hidden genetic variations can be detected with Chameleolyser, a new method developed in Nijmegen. The information is already yielding new patient diagnoses and may also lead to the discovery of as yet unknown disease genes, write Wouter Steyaert and Christian Gilissen of Radboudumc in Nature Communications.
Medical science has been using exome sequencing to map the genes of individual patients with rare diseases for about 15 years. With this technique, the DNA of a person's approximately 20,000 human genes are cut into small pieces so the DNA letters can be read off. This creates a huge amount of tiny DNA fragments, which are then reassembled into whole genes like a jigsaw puzzle. The result is an overview of that single person's 20,000 genes.
"Unfortunately, such an overview is never quite complete," says Christian Gilissen, professor of Genome Bioinformatics. "That's because of the evolution of our genome, our hereditary material. When copying DNA, things sometimes go wrong. Small pieces of DNA disappear or are added. Some pieces are copied more than once."
"It also happens that a copied gene is placed somewhere else in the genome, giving you a pseudogene in addition to the original gene. These genetic 'sloppinesses' are very important because they are the engine of evolution. This is how genetic changes arise. Changes that can be without effect or beneficial, but sometimes also cause new diseases."
Confusing pseudogenes
Zooming in on the gene and pseudogenes for a moment. The gene has a function, the pseudogene usually does not. Over time, small changes, mutations, can occur in both the gene and pseudogene. But gene and pseudogene are so similar that when sequencing it is not clear which piece belongs to the gene and which to the pseudogene.
Gilissen says, "For this reason, these DNA regions are not included in the analysis. A mutation found may originate from the pseudogene and have no meaning. If you add that mutation to the normal gene, you would make a wrong diagnosis. We don't want that."
Making the invisible visible
With Wouter Steyaert, Gilissen developed a method—Chameleolyser—that detects gene and pseudogene combinations in existing exome sequencing data and can also visualize the genetic variations between them.
Steyaert says, "We are now picking up a lot of genetic variation that was previously invisible. Per exome, we find about sixty additional genetic variants. For a number of people, this data allowed us to definitively determine the cause of their disease. With a new sequencing technique from PacBio, which analyzes longer stretches of DNA, we have established the reliability of our method."
The new method is interesting because it can be applied to already existing exome sequencing data. Thus, no new studies in patients are necessary. Any sequencing center in the world can apply the method.
"Such a large-scale analysis can also provide new biological insights," Gilissen says. "In many disorders, the genetic cause can only be determined in half of the patients. We think we will also find new disease genes in those gene-pseudogene combinations. For some of those patients, that may be where the genetic cause for their condition lies."
More information: Wouter Steyaert et al, Systematic analysis of paralogous regions in 41,755 exomes uncovers clinically relevant variation, Nature Communications (2023). DOI: 10.1038/s41467-023-42531-9