Protein creates paths for growing nerve cells
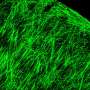
. Axons in mice without dystroglycan (right) are tangled and disorganized. Credit: Kevin Wright")
Working with mice, Johns Hopkins scientists have discovered that a particular protein helps nerve cells extend themselves along the spinal cord during mammalian development. Their results shed light on the subset of muscular dystrophies that result from mutations in the gene that holds the code for the protein, called dystroglycan, and also show how the nerve and muscle failings of the degenerative diseases are related.
As mammals like mice and humans develop, nerve cells in the brain and spinal cord must form connections with themselves and with muscles to assure proper control of movement. Nerve cells sometimes extend the whole length of the spinal cord to connect sensory nerves bearing information, for example, from the legs to the brain. To do so, nerve cells anchor their "headquarters," or cell bodies, in one location, and then extend a long, thin projection all the way to their target locations. These projections, or axons, can be 10,000 times longer than the cell body.
In a report published in the journal Neuron on Dec. 6, the authors suggest that, during fetal development, axons extend themselves along specific pathways created by dystroglycan.
"Dystroglycan creates a path and then binds other molecules that act as traffic signs, providing directional cues for axons en route to their targets," says David Ginty, Ph.D., a Howard Hughes investigator and professor of neuroscience in the Institute for Basic Biomedical Sciences at the Johns Hopkins University School of Medicine."
To find new genes associated with axon migration, Kevin Wright, Ph.D., a postdoctoral fellow in Ginty's laboratory, chemically provoked random genetic changes throughout the genomes of mice and then examined their axons for defective organization. What stood out were mutations in two genes that resulted in axon growth patterns similar to those in mice lacking the protein Slit, which is known to provide directional cues for nerve axons.
Digging deeper, Wright found that the mutated genes encode for the proteins B3gnt1 and ISPD, both of which had recently been implicated in adding sugar molecules to dystroglycan. Sugars bound to proteins often control their activity, so B3gnt1 and ISPD led the team to focus on dystroglycan as the key to the axon flaws they saw.
Through a series of genetic and biochemical experiments, the scientists learned that B3gnt1 and ISPD are indeed required for dystroglycan to work properly. Dystroglycan is made and released by specific cells within the spinal cord. It acts as a hub for tethering instructional molecules, like Slit, that guide nerve axons as they grow. But before dystroglycan can bind the instructional molecules, B3gnt1 and ISPD must attach sugars to it.
As expected, mice with defective dystroglycan usually have symptoms similar to mice with defective B3gnt1, ISPD or Slit. Moreover, the development of their muscles and nerves is severely disturbed, and the animals die soon after birth. Mouse fetuses completely missing any of the four proteins usually die before they are born.
Without dystroglycan or its helpers, the axons don't know where to go and the connection between nerve cells is lost, Ginty says. "Dystroglycan creates the road, B3gnt1 and ISPD add tar on top, and directional molecules like Slit stick to the tar and point the axons in the right direction," he explains. "If any part of the collaboration is missing, the axons overshoot their targets or don't turn where they should."
"Many muscular dystrophies are caused by a defect specific to muscles," says Ginty. "Children with defects in dystroglycan suffer from muscle weakening and bad nerve connections. Now we have mice with similar symptoms that we can study further."
More information: www.cell.com/neuron/abstract/S0896-6273(12)00899-9