Scientists uncover new details of natural anticancer mechanism
Scientists at The Scripps Research Institute (TSRI) have identified key triggers of an important cancer-blocking mechanism in cells.
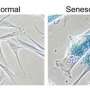
Termed "oncogene-induced senescence," this mechanism can block most cancer types, and is commonly experienced when incipient skin cancers turn instead into slow-growing moles. Tumors that achieve malignancy often do so by defeating or circumventing this growth barrier—which is why scientists have been eager to find out precisely how it works.
"We have known about some of the molecular signals that mediate this senescence response, but we've needed to understand the signaling pathway in much more detail," said Peiqing Sun, associate professor in TSRI's Department of Cell and Molecular Biology.
In the new study, published recently by the journal Molecular Cell, Sun and his colleagues describe the cascading interactions of three enzymes that are necessary to initiate a common type of oncogene-induced senescence.
Looking for Binding Partners
Oncogenes are growth-related genes that, through DNA damage, inherited mutations or some other cause, push cells to keep dividing beyond normal limits. Oncogenes in the ras gene family are the ones that have been most commonly linked to human cancers—and most studied as triggers of senescence.
Sun and other researchers showed a decade ago that an enzyme called p38 sits near the top of the ras-induced senescence response cascade. In 2007, Sun and his colleagues reported that p38 plays a role in this cascade by activating another enzyme, PRAK, through the addition of a phosphor group, a modification known as phosphorylation. For the new study, Sun and first author Research Associate Hui Zheng, along with other members of the laboratory, sought more details of PRAK's role in this cascade.
Zheng began the investigation by searching for binding partners of PRAK. With a series of protein-interaction assays he isolated an enzyme called Tip60, which binds tightly to PRAK. Further tests indicated that Tip60 does indeed lie within the senescence-inducing signaling cascade, because senescence fails to occur when Tip60 is absent.
PRAK is a kinase enzyme that, like p38, phosphorylates other proteins. Initially Zheng and Sun suspected that PRAK interacts with Tip60 by phosphorylating it, and thereby activating it.
Instead, the reverse turned out to be true: Tip60 acts on PRAK. Tip60 is a type of enzyme called an acetyltransferase, which modifies other proteins by adding acetyl groups. "Our tests showed that Tip60 binds to PRAK and acetylates it at a certain location, which helps activate PRAK," said Zheng.
Thus, the key enzyme PRAK requires two signals: "First the phosphorylation by p38 and then the acetylation by Tip60 are required for fully activating PRAK in this senescence–induction cascade," Zheng said.
Potential Cancer-Drug Strategy
What controls Tip60's own activation in this cascade? None other than the master switch, p38. "As a first step, p38 phosphorylates both Tip60 and PRAK," said Sun. Activated Tip60 then acetylates PRAK, completing PRAK's activation.
Previously Sun and his laboratory have shown that PRAK, when activated, goes on to activate the key tumor-suppressor protein p53, which exerts more direct control over a cell's growth machinery.
Sun and his team have been looking for ways to force the activation of the senescence response in cancer cells, as a potential cancer-drug strategy.
"Finding these details of the early part of the signaling cascade helps us understand better what we need to target," he said.
More information: "A Posttranslational Modification Cascade Involving p38, Tip60 and PRAK Mediates Oncogene-Induced Senescence," www.cell.com/molecular-cell/ab … 1097-2765(13)00294-3