Scientists define cellular pathway essential to removing damaged mitochondria
In a joint research effort with researchers at St. Jude Children's Research Hospital, and with help from scientists at The University of Pennsylvania, The University of Minnesota, and the National Institutes of Health, investigators from the Florida campus of The Scripps Research Institute have defined a specific protein complex that allows cells to rid themselves of damaged mitochondria, which are the energy producing machines of the cell.
"This protein complex is already being targeted in cancer therapeutics," said John Cleveland, chair of the Department of Cancer Biology at Scripps Florida, "but now we understand why some of the therapies that target this complex work and this new knowledge will have tremendous impact on both current and potential cancer therapies."
In particular, the study, which appears in the current issue of the journal Molecular Cell, focuses on how the cell uses a process known as autophagy—the major recycling center of the cell—to remove damaged mitochondria. The autophagy pathway is exploited by many tumors to survive stressful conditions and to remove damaged components.
The Cell under Stress
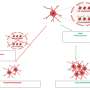
On a molecular level, the new study focuses on the role of the molecular complex known as "Hsp90-Cdc37 chaperone complex," which orchestrates various aspects of the cellular stress response. Although scientists had known that both the Hsp90-Cdc37 complex and autophagy help maintain the integrity of mitochondria, the exact relationship between Hsp90-Cdc37 and autophagy has not been well understood until the new study.
Hsp90, is a heat-shock protein, one of the cell's most abundant proteins, and assists in everything from protein folding and tumor repression to cell signaling. Cdc37, also a protein, is a co-chaperone to Hsp90 and is involved in cell signal transduction and connecting Hsp90 to the right kinases (kinases add a phosphate group to various molecules and can modify protein activity).
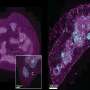
The study highlights the interaction between Hsp90-Cdc37 and Ulk1, a kinase that the authors show is required for the degradation and elimination of damaged mitochondria. Hsp90-Cdc37 stabilizes and activates Ulk1, which in turn phosphorylates its substrate Atg13, which is then released from the complex. Atg13 then eliminates damaged mitochondria via the autophagy pathway. Thus, the study links Hsp90-Cdc37-Ulk1-Atg13 in a direct pathway that is essential for efficient mitochondrial clearance.
"The new study shows that the key regulatory mechanism of this process is the Hsp90-Cdc37 chaperone, which functions as an on-off switch that is critical for the correct functioning of the Ulk1 kinase," Cleveland said. "Thus, if we can control this switch, we can significantly improve the therapeutic window."
More information: "Hsp90-Cdc37 Chaperone Complex RegulatesUlk1- and Atg13-mediated Mitophagy," www.cell.com/molecular-cell/abstract/S1097-2765%2811%2900464-3