Study of cox-2 inhibitors could lead to new class of stroke drugs
A study, in mice, by investigators at the Stanford University School of Medicine points toward potential new therapies for stroke, the nation's third-leading cause of death and foremost single cause of severe neurological disability. The study, which will be published online Oct. 3 in the Journal of Clinical Investigation, also may reveal why a much-heralded class of blockbuster drugs failed to live up to their promise.
Medical experts were excited when over a decade ago a class of drugs called COX-2-selective inhibitors came along. These new drugs were supposed to retain the advantages of aspirin and other so-called non-steroidal anti-inflammatory drugs, or NSAIDs, without causing stomach damage.
But in large-scale clinical trials of COX-2-selective inhibitors, puzzling — and disturbing — side effects emerged: namely, an increased risk of heart attacks and strokes. One already-approved drug in this category, rofecoxib (Vioxx), was pulled from the market in 2004. Another drug in this class is celecoxib, or Celebrex.
The new study helps explain why these drugs can be troublesome and how there may actually be some benefits to reap from the very molecular activity that these drugs were intended to block. "Some of what COX-2 does, it turns out, is good," said Katrin Andreasson, MD, associate professor of medicine and the study's senior author.
NSAIDs block both COX-2 and COX-1, two very similar versions of cyclo-oxygenase, an enzyme that catalyzes a key chemical reaction in the production of five related hormone-like messenger molecules called prostaglandins. The workings of COX-1 and COX-2 cause these five prostaglandin types to be produced in different ratios.
Prostaglandins travel from one cell to another, landing on and binding to dedicated receptor molecules sitting on cells' surfaces and stimulating various activities inside those cells. Each type of prostaglandin can trigger distinct effects. One prostaglandin in particular, PGE2, is known to be associated with pain and inflammation.
Because PGE2 is produced in relative abundance by COX-2 action, COX-2-selective inhibitors cause PGE2 levels to drop both absolutely and relative to the other prostaglandins. That makes them effective pain relievers. But Andreasson wanted to understand why they can cause strokes.
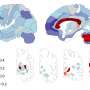
PGE2 has four separate counterpart receptors, designated EP1 through EP4, each of which sets in motion a different set of activities inside cells on binding to PGE2. Andreasson's team used a mouse model of stroke to show that activating one of these receptors, EP4, after a traumatic brain event such as a stroke can be very beneficial.
In the early 2000s, Andreasson carried out studies indicating that COX-2 activity is normally quite strong in nerve cells, where it appears to be involved in physiological changes underpinning learning. Now, in the new study, she and her colleagues found that amounts of EP4 on both nerve cells and the endothelial cells that line the blood vessels of the brain increase substantially after a stroke.
"This, to us, suggested that the EP4 receptor may be doing something important," she said.
Andreasson's team employed a compound that selectively binds to and activates EP4 to show that administering this compound by injection as much as three hours after a stroke reduced the amount of brain damage the mice suffered. Importantly, a single injection of this compound three hours after a stroke enhanced mice's behavioral recovery from the stroke, as measured a full week later by their superior performance on a test of motor coordination.
The researchers then examined the independent effects of EP4 activation on nerve cells and endothelial cells. Adding the compound increased the survival of nerve cells in a dish after the cells had been stressed by conditions similar to a stroke. In endothelial cells, EP4 activation by the compound resulted in an increased production of nitric oxide, a key chemical that among other things diffuses to nearby smooth muscle cells surrounding blood vessels, relaxing them. This lets the blood vessels dilate, enhancing blood flow.
Inactivation, via a sophisticated genetic manipulation, of the EP4 receptor on the mice's nerve cells both increased stroke severity in the mice and worsened their recovery. Similarly inactivating the receptor on endothelial cells worsened stroke injury and decreased blood flow to the affected area.
The only currently approved drug for stroke is tissue plasminogen activator, or tPA, which dissolves clots that prevent oxygenated blood from reaching brain tissue. But tPA does nothing to counter the damage caused after the stroke by inflammatory agents that flood into the affected tissue. New treatments for this debilitating disorder are desperately needed, Andreasson said. A drug that increased blood flow from still-functioning vasculature to the stroke region, as the EP4-activating compound does, might complement tPA's effect, she added.
"We showed that activating this single receptor, EP4, three hours after a stroke not only diminishes the volume of a mouse's affected brain tissue but also enhances the mouse's functional recovery," said Andreasson. "And we've taken this a step further by diligently unraveling the mechanisms by which that happens."
Andreasson's laboratory is now exploring the behavioral recovery of mice given the EP4-activating compound after longer post-stroke waiting times. She said she wants to see whether the therapeutic window can be extended to more than six hours after a stroke, which could greatly increase the value of such a treatment.
Andreasson cautioned that many therapies, even when highly successful in mice, fail in the clinic.