Study unravels central mystery of Alzheimer's disease
Scientists at The Scripps Research Institute (TSRI) have shed light on one of the major toxic mechanisms of Alzheimer's disease. The discoveries could lead to a much better understanding of the Alzheimer's process and how to prevent it.
The findings, reported in the April 10, 2013 issue of the journal Neuron, show that brain damage in Alzheimer's disease is linked to the overactivation of an enzyme called AMPK. When the scientists blocked this enzyme in mouse models of the disease, neurons were protected from loss of synapses—neuron-to-neuron connection points—typical of the early phase of Alzheimer's disease.
"These findings open up many new avenues of investigation, including the possibility of developing therapies that target the upstream mechanisms leading to AMPK overactivation in the brain," said TSRI Professor Franck Polleux, who led the new study.
Alzheimer's disease, a fatal neurodegenerative disorder afflicting more than 25 million people worldwide, currently has no cure or even disease-delaying therapy.
In addition to having implications for Alzheimer's drug discovery, Polleux noted the findings suggest the need for further safety studies on an existing drug, metformin. Metformin, a popular treatment for Type 2 Diabetes, causes AMPK activation.
Tantalizing Clues to Alzheimer's
Researchers have known for years that people in the earliest stages of Alzheimer's disease begin to lose synapses in certain memory-related brain areas. Small aggregates of the protein amyloid beta can cause this loss of synapses, but how they do so has been a mystery.
Until recently, Polleux's laboratory has been focused not on Alzheimer's research but on the normal development and growth of neurons. In 2011, he and his colleagues reported that AMPK overactivation by metformin, among other compounds, in animal models impaired the ability of neurons to grow output stalks, or axons.
Around the same time, separate research groups found clues that AMPK might also have a role in Alzheimer's disease. One group reported that AMPK can be activated in neurons by amyloid beta, which in turn can cause a modification of the protein tau in a process known as phosphorylation. Tangles of tau with multiple phosphorylations ("hyperphosphorylated" tau) are known to accumulate in neurons in affected brain areas in Alzheimer's. These results, published two years ago, reported abnormally high levels of activated AMPK in these tangle-ridden neurons.
Polleux decided to investigate further, to determine whether the reported interactions of AMPK with amyloid beta and tau can in fact cause the damage seen in the brains of Alzheimer's patients. "Very little was known about the function of this AMPK pathway in neurons, and we happened to have all the tools needed to study it," he said.
In Search of Answers
Georges Mairet-Coello, a postdoctoral research associate in the Polleux lab, performed most of the experiments for the new study. He began by confirming that amyloid beta, in the small-aggregate ("oligomer") form that is toxic to synapses, does indeed strongly activate AMPK; amyloid beta oligomers stimulate certain neuronal receptors, which in turn causes an influx of calcium ions into the neurons. He found that this calcium influx triggers the activation of an enzyme called CAMKK2, which appears to be the main activator of AMPK in neurons.
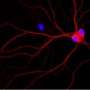
The team then showed that this AMPK overactivation in neurons is the essential reason for amyloid beta's synapse-harming effect. Normally, the addition of amyloid beta oligomers to a culture of neurons causes the swift disappearance of many of the neurons' dendritic spines—the rootlike, synapse-bearing input stalks that receive signals from other neurons. With a variety of tests, the scientists showed that amyloid beta oligomers can't cause this dendritic spine loss unless AMPK overactivation occurs—and indeed AMPK overactivation on its own can cause the spine loss.
For a key experiment the team used J20 mice, which are genetically engineered to overproduce mutant amyloid beta, and eventually develop an Alzheimer's-like condition. "When J20 mice are only three months old, they already show a strong decrease in dendritic spine density, in a set of memory-related neurons that are also affected early in human Alzheimer's," Mairet-Coello said. "But when we blocked the activity of CAMKK2 or AMPK in these neurons, we completely prevented the spine loss."
Next Mairet-Coello investigated the role of the tau protein. Ordinarily it serves as a structural element in neuronal axons, but in Alzheimer's it somehow becomes hyperphosphorylated and drifts into other neuronal areas, including dendrites where its presence is associated with spine loss. Recent studies have shown that amyloid beta's toxicity to dendritic spines depends largely on the presence of tau, but just how the two Alzheimer's proteins interact has been unclear.
The team took a cue from a 2004 study of Drosophila fruit flies, in which an AMPK-like enzyme's phosphorylation of specific sites on the tau protein led to a cascade of further phosphorylations and the degeneration of nerve cells. The scientists confirmed that one of these sites, S262, is indeed phosphorylated by AMPK. They then showed that this specific phosphorylation of tau accounts to a significant extent for amyloid beta's synapse toxicity. "Blocking the phosphorylation at S262, by using a mutant form of tau that can't be phosphorylated at that site, prevented amyloid beta's toxic effect on spine density," Mairet-Coello said.
The result suggests that amyloid beta contributes to Alzheimer's via AMPK, mostly as an enabler of tau's toxicity.
More Studies Ahead
Mairet-Coello, Polleux and their colleagues are now following up with further experiments to determine what other toxic processes, such as excessive autophagy, are promoted by AMPK overactivation and might also contribute to the long-term aspects of Alzheimer's disease progression. They are also interested in the long-term effects of blocking AMPK overactivation in the J20 mouse model as well as in other mouse models of Alzheimer's disease, which normally develop cognitive deficits at later stages. "We already have contacts within the pharmaceuticals industry who are potentially interested in targeting either CAMKK2 or AMPK," says Polleux.
The other contributors to the study, "The CAMKK2-AMPK kinase pathway mediates the synaptotoxic effects of amyloid beta oligomers through tau phosphorylation," were Julien Courchet, Simon Pieraut, Virginie Courchet and Anton Maximov, all of TSRI.