New therapeutic target discovered for Alzheimer's disease

A team of scientists from the University of California, San Diego School of Medicine, the Medical University of South Carolina and San Diego-based American Life Science Pharmaceuticals, Inc., report that cathepsin B gene knockout or its reduction by an enzyme inhibitor blocks creation of key neurotoxic pGlu-Aβ peptides linked to Alzheimer's disease (AD). Moreover, the candidate inhibitor drug has been shown to be safe in humans.
The findings, based on AD mouse models and published online in the Journal of Alzheimer's Disease, support continued development of cysteine protease inhibitors as a new drug target class for AD. "No other therapeutic program is investigating cysteine protease inhibitors for treating AD," said collaborator Vivian Hook, PhD, professor in the UC San Diego Skaggs School of Pharmacy and Pharmaceutical Sciences and in the UC San Diego School of Medicine.
Current AD drugs treat some symptoms of the devastating neurological disorder, but none actually slow its progress, prevent or cure it. No new AD drug has been approved in more than a decade.
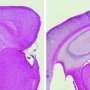
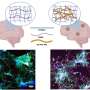
The researchers focused on cathepsin B production of N-truncated pGlu-Aβ, a peptide or short chain of amino acids, and the blockade of cathepsin B by E64d, a compound shown to inhibit cysteine proteases, a type of enzyme. AD is characterized by accumulation of a variety of Aβ peptides as oligomers and amyloid plaques in the brain, factors involved in neuronal loss and memory deficits over time. These neurotoxic Aβ peptides are created when enzymes cleave a large protein called amyloid precursor protein (APP) into smaller Aβ peptides of varying toxicity. N-truncated pGlu-Aβ has been shown to be among the most neurotoxic of multiple forms of Aβ peptides.
Much AD research has focused on the APP-cutting enzyme BACE1 β-secretase, but its role in producing pGlu-Aβ was unknown. Cathepsin B is an alternative β-secretase which cleaves the wild-type β-secretase site of APP, which is expressed in the major sporadic and many familial forms of AD. Hook and colleagues looked at what happened after gene knockout of BACE1 or cathepsin B. They found that cathepsin B, but not BACE1, produced the highly toxic pGlu-Aβ.
Perhaps most interestingly, the scientists found that E64d, an enzyme inhibitor of cathepsin B, reduced production of pGlu-Aβ and other AD-associated Aβ peptides. Key was the finding that E64d and cathepsin B gene knock out resulted in improved memory deficits in a mouse model of AD.
"This is an exciting finding," said Hook. "It addresses a new target – cathepsin B – and an effective, safe small molecule, E64d, to reduce the pGlu-Aβ that initiates development of the disease's neurotoxicity. No other work in the field has addressed protease inhibition for reducing pGlu-Aβ of AD."
Hook noted that E64d has already been shown to be safe in clinical trials of patients with muscular dystrophy and would, therefore, likely prove safe for treating AD as well. She hopes to launch Phase 1 human clinical trials in the near future with a modified version of the drug candidate.