Mutations in key cancer protein suggest new route to treatments
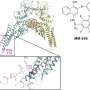
. Previously, efforts to interfere with STAT3’s contribution to cancer have focused on the SH2 domains (yellow) at the tip of the pliers. Darnell and his team instead altered the linker domains (orange).")
For years, scientists have struggled to find a way to block a protein known to play an important role in many cancers. The protein, STAT3, acts as a transcription factor—it performs the crucial task of helping convert DNA into the RNA instructions used to make new proteins.
But when overly active, STAT3 performs this task too well, fueling the growth and division of abnormal cells, and contributing to cancer. Scientists have taken various approaches to selectively blocking STAT3 in cancer, but none have produced successful treatments.
Now, researchers led by Rockefeller University's James E. Darnell, head of the Laboratory of Molecular Cell Biology, have suggested a new way to target STAT3. In research published November 9 in the Proceedings of the National Academy of Sciences, they report successfully disrupting STAT3's ability to act as a transcription factor, suggesting a basis for new, targeted approaches to fighting cancer.
"We have described some interesting mutations in the STAT3 protein that, if we could mimic with a drug, could become very valuable tools in our fight against cancer," says Darnell, Vincent Astor Professor Emeritus at Rockefeller. "Some of the mutations, in particular, seem really exciting."
Many scientists—and drug companies—have focused on STAT3 because it is overactive in virtually all of the major human cancers: breast, prostate, lung, colon, and some blood malignancies. But earlier efforts have not succeeded in finding drugs that block the protein at low enough doses, Darnell says.
In order to do its normal job, an individual STAT3 is activated by addition of an electrically charged chemical group and then must pair with another STAT3, forming what's known as a dimer that adopts a particular configuration. "The shape is like an open pair of pliers grabbing a wire, where the wire represents DNA," says Darnell.
The points that touch the DNA—sparking the process of converting its code into protein—are known as the DNA-binding domains. At the tip of the pliers is the SH2 domain, which keeps the two charged STAT3s together. It's the SH2 domain that most scientists have focused on, says Darnell, because they believed that by blocking this domain, the two halves of the dimer would fall apart, and STAT3 couldn't do its job. But despite many attempts, scientists have yet to find a way to fully block the SH2 interactions, he notes.
So, Darnell and the other researchers, including first author Claudia Mertens, a senior research associate in the lab, decided to approach the problem in a different way. What if, instead of trying to disable the domain that bound the two STAT3s together as a dimer, they focused on another region of the STAT3 structure entirely? The linker domain, which connects the SH2 and DNA-binding domains, particularly interested them because of earlier work in the lab. Interfering with this domain of STAT3, they reasoned, could theoretically change either of the other two domains, since it comes in contact with both.
Darnell and his colleagues say their work shows that a number of mutations in the linker domain of STAT3 appear to block the action of the transcription factor. Co-author Sebastian Klinge, assistant professor at Rockefeller and head of the Laboratory of Protein and Nucleic Acid Chemistry, helped the team identify the likely effects of the mutations on the STAT3 structure.
"Three of the mutations, for instance, enable the STAT3 dimer to bind to DNA, but prevent the transcription of that DNA into RNA instructions for protein synthesis, a crucial step for cancer cells," says Darnell.
The findings help reinforce the importance of targeting transcription factors such as STAT3 in the fight against cancer, Darnell notes. "What's the good of knowing the intricacies of how proteins work if we can't use that information to stop cancer in a targeted way?"
More information: C. Mertens et al. Mutations in the linker domain affect phospho-STAT3 function and suggest targets for interrupting STAT3 activity, Proceedings of the National Academy of Sciences (2015). DOI: 10.1073/pnas.1515876112