Existing compound holds promise for reducing Huntington's disease progression
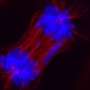
. The neuron in the center (yellow) contains an abnormal intracellular accumulation of huntingtin called an inclusion body (orange). Credit: Wikipedia/ Creative Commons Attribution 3.0 Unported license")
Currently, there is no treatment to halt the progression of Huntington's disease (HD), a fatal genetic disorder that slowly robs sufferers of their physical and mental abilities. Now, researchers at University of California, San Diego School of Medicine have discovered that an existing compound, previously tested for diabetes, offers hope for slowing HD and its symptoms.
The study is published in the December 7, 2015 online issue of Nature Medicine.
"We're very excited by our pre-clinical testing of this compound (KD3010)," said Albert La Spada, MD, PhD, professor of pediatrics, cellular and molecular medicine and neurosciences at UC San Diego School of Medicine. "It improved motor function, reduced neurodegeneration and increased survival in a mouse model of Huntington's disease and reduced toxicity in neurons generated from human HD stem cells."
La Spada said these findings are particularly notable because the drug was well-tolerated by participants in a Phase Ib clinical trial for diabetes, conducted in 2006, by a now-defunct biotech company.
"We have a drug that was FDA approved for human use in a clinical trial. It did not produce any significant side effects. This means it has a good safety profile and so likely can be moved into testing in Huntington's patients much more quickly than compounds that have not been tested in humans. This is important since right now there is zero that can be done to alter the progression of this devastating disease."
La Spada's discovery of the drug's potential in HD builds upon more than a decade of research into the disorder's underlying molecular pathology. Much of that work has centered on misfolded proteins, which are known to be key culprits in HD and several other neurodegenerative diseases.
In HD, an inherited mutation in the huntingtin (htt) gene results in misfolded htt proteins accumulating in certain central nervous system cells, leading to progressive deterioration of involuntary movement control, cognitive decline and psychological problems. Approximately 30,000 Americans have HD and more than 200,000 are at-risk of inheriting the disease.
La Spada, also chief of the Division of Genetics in the Department of Pediatrics at UC San Diego School of Medicine, is now consulting with other HD researchers about clinical trial design and hopes to initiate a KD3010 trial within 18 months.
Over the years, La Spada and his lab have sought to illuminate the cellular steps triggered by the mutant htt gene, in hopes of fully understanding how the gene mutation causes HD's progressive breakdown of nerve cells in the brain.
In a major finding in 2006, La Spada and others showed that the mutated htt gene interferes with the function of PGC-1 alpha, a regulatory protein central to the creation and operation of mitochondria, the cell's energy factory. "By disrupting this function, the mutant htt gene reduces energy function needed for a nerve cell's survival," said La Spada.
In 2012, he expanded on PGC-1 alpha's role in HD with the discovery that its disruption also undermined healthy brain cells' ability to degrade damaged molecules. This leads to the accumulation of the problematic misfolded proteins, a hallmark of Huntington's, said La Spada. Conversely, he found that elevating levels of PGC-1 alpha in HD mice virtually eliminated the misfolded proteins and rescued neurological disease and neurodegeneration in the HD mice.
In his latest study, La Spada delved deeper into reduced PGC-1 alpha activity by evaluating certain key transcription factors that rely upon PGC-1 alpha for their ability to function. La Spada and colleagues noted that one of them, peroxisome proliferator-activated receptor delta (PPARδ), physically interacted with the htt protein in the brains of HD mice, and its function was greatly impaired in HD nerve cells. PPARδ had already been studied extensively in skeletal muscle, where it was found to enhance physical activity. "We learned that PPARδ was expressed in even larger amounts in the brain and we began to suspect that it might be important for brain function," said La Spada.
The researchers bred mice with a mutant version of PPARδ that would turn off, thereby reducing energy production and degradation of misfolded proteins in nerve cells. "These mice developed very severe symptoms of Huntington's disease," said La Spada. "This showed us that PPARδ was needed to make sure neurons worked optimally."
About the same time, La Spada became aware of a drug - KD3010 - developed a decade before and tested in diabetes trials, that would increase PPARδ activity. Last year, La Spada's team synthesized the compound and began testing it in HD mice. "We saw significant improvements in terms of neurodegeneration," he said. "It also extended their lives by 16 percent."
At the cellular level, La Spada said the drug improved mitochondrial energy production and helped mice get rid of the misfolded proteins. Since misfolded proteins also underlie Alzheimer's, Parkinson's and other neurodegenerative disorders, La Spada hopes that, if successful in HD, the compound can also be tested in other related neurological diseases.
More information: PPAR-δ is repressed in Huntington's disease, is required for normal neuronal function and can be targeted therapeutically, Nature Medicine, DOI: 10.1038/nm.4003