New therapy halts progression of Lou Gehrig's disease in mice
")
Researchers at Oregon State University announced today that they have essentially stopped the progression of amyotrophic lateral sclerosis (ALS), or Lou Gehrig's disease, for nearly two years in one type of mouse model used to study the disease - allowing the mice to approach their normal lifespan.
The findings, scientists indicate, are some of the most compelling ever produced in the search for a therapy for ALS, a debilitating and fatal disease, and were just published in Neurobiology of Disease.
"We are shocked at how well this treatment can stop the progression of ALS," said Joseph Beckman, lead author on this study, a distinguished professor of biochemistry and biophysics in the College of Science at Oregon State University, and principal investigator and holder of the Burgess and Elizabeth Jamieson Chair in OSU's Linus Pauling Institute.
In decades of work, no treatment has been discovered for ALS that can do anything but prolong human survival less than a month. The mouse model used in this study is one that scientists believe may more closely resemble the human reaction to this treatment, which consists of a compound called copper-ATSM.
It's not yet known if humans will have the same response, but researchers are moving as quickly as possible toward human clinical trials, testing first for safety and then efficacy of the new approach.
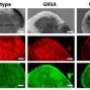
ALS was identified as a progressive and fatal neurodegenerative disease in the late 1800s, and gained international recognition in 1939 when it was diagnosed in American baseball legend Lou Gehrig. It's known to be caused by the death and deterioration of motor neurons in the spinal cord, which in turn has been linked to mutations in copper, zinc superoxide dismutase.
Copper-ATSM is a known compound that helps deliver copper specifically to cells with damaged mitochondria, and reaches the spinal cord where it's needed to treat ALS. This compound has low toxicity, easily penetrates the blood-brain barrier, is already used in human medicine at much lower doses for some purposes, and is well tolerated in laboratory animals at far higher levels. Any copper not needed after use of copper-ATSM is quickly flushed out of the body.
Experts caution, however, that this approach is not as simple as taking a nutritional supplement of copper, which can be toxic at even moderate doses. Such supplements would be of no value to people with ALS, they said.
The new findings were reported by scientists from OSU; the University of Melbourne in Australia; University of Texas Southwestern; University of Central Florida; and the Pasteur Institute of Montevideo in Uruguay. The study is available as open access in Neurobiology of Disease.
Using the new treatment, researchers were able to stop the progression of ALS in one type of transgenic mouse model, which ordinarily would die within two weeks without treatment. Some of these mice have survived for more than 650 days, 500 days longer than any previous research has been able to achieve.
In some experiments, the treatment was begun, and then withheld. In this circumstance the mice began to show ALS symptoms within two months after treatment was stopped, and would die within another month. But if treatment was resumed, the mice gained weight, progression of the disease once again was stopped, and the mice lived another 6-12 months.
In 2012, Beckman was recognized as the leading medical researcher in Oregon, with the Discovery Award from the Medical Research Foundation of Oregon. He is also director of OSU's Environmental Health Sciences Center, funded by the National Institutes of Health to support research on the role of the environment in causing disease.
"We have a solid understanding of why the treatment works in the mice, and we predict it should work in both familial and possibly sporadic human patients," Beckman said. "But we won't know until we try."
Familial ALS patients are those with more of a family history of the disease, while sporadic patients reflect the larger general population.
"We want people to understand that we are moving to human trials as quickly as we can," Beckman said. "In humans who develop ALS, the average time from onset to death is only three to four years."
The advances are based on substantial scientific progress in understanding the disease processes of ALS and basic research in biochemistry. The transgenic mice used in these studies have been engineered to carry the human gene for "copper chaperone for superoxide dismutase," or CCS gene. CCS inserts copper into superoxide dismustase, or SOD, and transgenic mice carrying these human genes die rapidly without treatment.
After years of research, scientists have developed an approach to treating ALS that's based on bringing copper into specific cells in the spinal cord and mitochondria weakened by copper deficiency. Copper is a metal that helps to stabilize SOD, an antioxidant protein whose proper function is essential to life. But when it lacks its metal co-factors, SOD can "unfold" and become toxic, leading to the death of motor neurons.
There's some evidence that this approach, which works in part by improving mitochondrial function, may also have value in Parkinson's disease and other conditions, researchers said. Research is progressing on those topics as well.
The treatment is unlikely to allow significant recovery from neuronal loss already caused by ALS, the scientists said, but could slow further disease progression when started after diagnosis. It could also potentially treat carriers of SOD mutant genes that cause ALS.
More information: Jared R. Williams et al. Copper delivery to the CNS by CuATSM effectively treats motor neuron disease in SODG93A mice co-expressing the copper-chaperone-for-SOD, Neurobiology of Disease (2016). DOI: 10.1016/j.nbd.2016.01.020